制备5-羟基烷基-取代的1-苯基-1,2,4-三唑衍生物的方法与流程
其中
r1a和r1b彼此独立地选自氢、氟、氯、甲基、单氟甲基、二氟甲基、三氟甲基、乙基、甲氧基、二氟甲氧基和三氟甲氧基,
涉及用于制备其的新型前体,和涉及式(i-a-1)的(5-(4-氯苯基)-2-({1-(3-氯苯基)-5-[(1s)-1-羟基乙基]-1h-1,2,4-三唑-3-基}甲基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮的结晶多形体i的制备和用途。
式(i)的化合物充当有效的双重v1a/v2受体拮抗剂,并可用作预防和/或治疗心血管疾病和肾疾,例如急性和慢性心力衰竭(恶化慢性心力衰竭)、心肾综合征、高容量性和低容量性低钠血症、肝硬化、腹水、水肿和adh分泌异常综合征(siadh)的药剂,如wo2016/071212中所公开。
制备5-苯基-取代的1,2,4-三唑衍生物的一般方法描述于wo2011/104322中(参见其中的图式8,实施例21、25、54、56-61和68-70)。然而,通过其中描述的方法,不可能在一个工艺步骤中实现1,2,4-三唑环的1,3,5取代,尤其是1,2,4-三唑环的1-苯基取代。
下面的图式1显示了根据wo2011/104322制备5-苯基取代的1,2,4-三唑衍生物的方法。
图式1:根据wo2011/104322合成1,2,4-三唑衍生物
[wo2011/104322:图式8,第32页;l2=尤其是键;ar2=尤其是取代的苯基;alk=烷基]。
式(i)的化合物及其制备描述于wo2016/071212中。其中描述的研究合成被认为是最接近的现有技术。由5-(4-氯苯基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(ii)起始,在4个阶段中制备式(i)的目标化合物,其中总收率为理论值的最多~12%。式(i-a)和(i-b)的非对映异构体在实验室规模上在进一步的阶段中通过手性非对映异构体分离由非对映异构体混合物(i)获得。
虽然式(i)的化合物在wo2016/071212中以固体形式获得,但迄今为止尚未描述制备制药可用晶形的最终阶段的特定结晶方法。
下面的图式2显示了制备式(i)的化合物的方法。
图式2:根据wo2016/071212合成式(i)的化合物
[r1a、r1b=氢、氟、氯、氰基、甲基、单氟甲基、二氟甲基、三氟甲基、乙基、甲氧基、二氟甲氧基和三氟甲氧基]。
直至并包括步骤(v)至(vii),wo2016/071212中公开的合成类似于wo2011/104322中公开的方法。
为了制备式(iv)的化合物,使5-(4-氯苯基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(ii)与溴乙酸甲酯(iii)反应以产生{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙酸甲酯(iv)。随后,使(iv)与水合肼转化成2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙酰肼(v)。然后使(v)与酰亚胺化合物(vi)反应以产生非对映异构体混合物5-(4-氯苯基)-2-({5-[(1rs)-1-羟基乙基]-1h-1,2,4-三唑-3-基}甲基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(vii)。通过(vii)与取代的苯基硼酸(viii)的经铜催化的芳基偶联(“chan-lam偶联”),产生取代的5-(1-羟基乙基)-1-芳基-1,2,4-三唑衍生物(i)。通过手性色谱法的分离,得到各自的非对映异构体5-(4-氯苯基)-2-({5-[(1s)-1-羟基乙基]-1-(r1a,r1b)-苯基-1h-1,2,4-三唑-3-基}甲基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(i-a)和5-(4-氯苯基)-2-({5-[(1r)-1-羟基乙基]-1-(r1a,r1b)-苯基-1h-1,2,4-三唑-3-基}甲基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(i-b)。
上述概述的反应图式在wo2016/071212中描述如下:式(ii)化合物经由式(iv)、(v)和(vii)的化合物产生式(i)的化合物的反应序列以及分离成非对映异构体(i-a)和(i-b)参见其中的图式2和实施例1a、2a、4a和10至83。
然而,从wo2016/071212中已知的该方法在反应方案中具有各种缺点,其在工业规模中制备式(i)的化合物中具有特别不利的影响。四个阶段(ii)至(i)的总收率非常低,其低于理论值的15%(约1.3%至13.1%)。许多步骤以非常高的稀释度和非常高的试剂过量进行。
发现wo2016/071212中描述的合成的特别不利方面是合成步骤(vii)至(i)(铜介导的芳基偶联,“chan-lam偶联”),其仅以最大30%的分离收率(3.0%至30.1%)进行,因此从原子经济性的观点来看是不利的。另一个缺点是在该反应中可以经由与另一个环氮原子的偶联反应(1,2,4-三唑衍生物(vii)的环互变异构)产生区域异构的苯基三唑衍生物。这同样对该步骤中的收率具有不利影响;此外,然后必须在额外的提纯步骤中以复杂的方式分离出所述区域异构产物。此外,该反应步骤对于工业规模的合成特别不利,因为在该反应中使用化学计算量的乙酸铜。这是不利的,因为必须将剩余量的铜盐除去直至低于在每种情况中在产品中出于法规原因而允许的最大限值,这意味着额外的支出。而且,试剂应该以简单和廉价的方式可得。
在根据wo2016/071212的合成中,通过手性色谱法在实验室规模上将式(i)的立体异构混合物分离成非对映异构体。这种色谱分离非常昂贵且耗时,因此对于工业规模的合成是不利的。此外,由于这一额外阶段,进一步降低了总收率。另一个缺点是,通过wo2016/071212中所述方法的目标化合物不以制药可用的特定晶形获得。
因此,需要一种在工业规模上可行的合成方法,其可再现地以高的总收率、低生产成本和高纯度提供式(i)的化合物。此外还需要一种在工业规模上可行的合成方法,其满足活性物质用于临床试验中且可用于后续的官方提交所要遵守的所有法规要求。
令人惊奇的是,现已发现一种制备式(i)的化合物的非常有效的方法,其满足上述要求。该新方法能够有效合成5-羟基烷基-取代的1-苯基-1,2,4-三唑衍生物。
本发明方法的一个重要优点是通过所有阶段明显增加的收率。根据方法变体(a)的本发明新型方法在四个阶段中以大于理论值的20%(23.8%至53.2%)的总收率提供目标化合物(i)。中间体的色谱提纯是不必要的。因此,替代性方法变体(b)中的最后两个阶段,另一个替代性方法变体(c)中的甚至最后三个阶段可以作为一锅法进行。通过这种方式,可以实现通过四个阶段的总收率的进一步增加(高达63.2%)。
下文描述的图式和方法步骤是通式(i)的本发明化合物的合成路线,且不应视为限制。本领域技术人员知道,可以以多样化方式修改如图式3和4中的示例性展示的转化序列,因此所展示的序列不应被视为限制。此外,可以在示例性描述的转化之前和/或之后转化各个基团和取代基的官能团,尤其是在r1和r2下列出的那些,其中由通过上述方法获得的其它式(i)的化合物或其前体起始。这些转化通过本领域技术人员熟悉的常规方法进行,例如包括反应,如亲核或亲电取代反应、过渡金属介导的偶联反应、金属有机化合物(例如格氏化合物或锂有机化合物)的制备和加成反应、氧化和还原反应、氢化、卤化(例如氟化、溴化)、脱卤、胺化、烷基化和酰化、形成羧酸酯、羧酰胺和磺酰胺、酯裂解和水解以及引入和除去临时保护基或本领域技术人员已知的其它反应。这些转化还包括其中引入能够进一步转化取代基的官能的那些。合适的保护基和用于引入和脱除其的试剂和反应条件是本领域技术人员已知的(参见例如t.w.greene和p.g.m.wuts;"protectivegroupsinorganicsynthesis”,第3版,wiley1999)。在下面的文本段落中列出具体实例。
下面的图式3说明了制备式(i)的化合物的本发明新方法。
图式3:制备式(i)的化合物的本发明方法
[r1a、r1b=氢、氟、氯、甲基、单氟甲基、二氟甲基、三氟甲基、乙基、甲氧基、二氟甲氧基和三氟甲氧基;a)na2co3、甲基异丁基酮;b)甲醇钠、meoh,c)1.(xii-a)、dipea、甲苯/thf,2.(xiii)、dipea、thf;d)naoh、meoh]。
其中,在合成图式3的化合物中,
r1a和r1b彼此独立地选自氢、氟、氯、甲基、单氟甲基、二氟甲基、三氟甲基、乙基、甲氧基、二氟甲氧基和三氟甲氧基。
其中,优选地,在合成图式3的化合物中,
r1a和r1b彼此独立地选自氢、氟和氯,其中所述取代基的至少之一不是氢。
其中,更优选地,在合成图式3的化合物中,
r1a是氢,且
r1b在2位或3位是氯。
其中,最优选地,在合成图式3的化合物中,
r1a是氢,且
r1b在3位是氯。
下面根据图式3讨论制备式(i)的化合物的本发明方法的各个阶段。同样讨论了替代性方案,其特征在于式(xi)和(xiv)的化合物未分离。
为了制备5-(1-羟基乙基)-1-芳基-1,2,4-三唑衍生物(i),使5-(4-氯苯基)-4-((2s)-3,3,3-三氟-2-羟基丙基)-2,4-二氢-3h-1,2,4-三唑-3-酮(ii)转化成{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙腈(x)(步骤1)。随后,使腈化合物(x)通过与甲醇钠反应而转化成2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙亚氨酸甲酯(xi)(步骤2)。然后经由三组分环化反应形成1,2,4-三唑环,其中亚氨酸酯化合物(xi)与2-乙酰氧基丙酰氯(xii)和取代的苯基肼化合物(xiii)反应,并且获得乙酸1-{3-({3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}甲基)-1-[(r1a,r1b)-苯基]-1h-1,2,4-三唑-5-基}乙基酯(xiv)(步骤3)。随后脱除乙酰基,提供式(i)的目标化合物(步骤4)。在一个变体(b)中,该方法可以如下方式进行,以使得受保护的乙酸酯(xiv)不经分离,而是在溶液中直接进一步转化(步骤3+4)。在另一个变体(c)中,该方法可以经由阶段(x)→(xi)→(xiv)→(i)作为一锅法进行(步骤2+3+4);在这种情况下,整个方法仅由2个分开的阶段而非现有技术中的4个阶段构成。
特别有利的是根据本发明的用于形成1,2,4-三唑环的三组分环化反应(步骤3),其能够在一个方法步骤中在第1和5位引入两个环取代基,以使得该步骤达到高收率(步骤3为37.0%至83.0%;脱除保护基后:步骤3+4通过2阶段为36.7%至82.2%)。现有技术中描述的合成(图式2)经由类似序列(v)→(vii)→(i)产生通过两个阶段的2.9%至28.9%的显著更低的收率。
此外,有利的还是本发明方法的稳健性,以使得如上所述,序列也可以作为一锅法(方法变体b和c)进行,以使得中间体不需要经分离,并且不通过色谱法提纯中间体,而是使其在相同的反应容器和/或反应介质中直接进行后续步骤。这种一锅法对于工业规模合成是特别有利的,因为以这种方式可以避免额外的后处理步骤,并且该方法实现高的总收率。
合成图式3中描述的式(ii)的起始化合物可以根据下面的合成图式4由可商购的或本领域技术人员已知的起始化合物制备:
图式4:制备式(ii)的化合物的方法
[e)thf;f)naoh水溶液、δ;g)1.(cf3co)2o/吡啶,2.hcl水溶液、δ;h)手性ru(ii)催化剂、hcooh/et3n]。
式(ii)的起始物质描述于wo2010/105770(参见其中图式4和5;实施例1a、2a、3a、4a和158a)和wo2011/104322(参见其中图式1;其实施例1a、2a、3a、4a和5a)。式(ii)的化合物通过使4-氯苯甲酰肼(xv)与2-异氰酸根合乙酸乙酯(xvi)反应产生n-({2-[(4-氯苯基)羰基]肼基}羰基)甘氨酸乙酯(xvi)而获得。然后将其通过碱诱导的环化反应转化成[[3-(4-氯苯基)-5-氧代-1,5-二氢-4h-1,2,4-三唑-4-基]乙酸(xviii)。5-(4-氯苯基)-4-(3,3,3-三氟-2-氧代丙基)-2,4-二氢-3h-1,2,4-三唑-3-酮(ix)然后通过与三氟乙酸酐反应和随后用盐酸处理而获得。然后借助对映选择性钌(ii)催化剂通过不对称氢转移将酮(ix)转化成手性醇(ii)。
利用新型的本发明合成,可以以非常有效的方式制备目标化合物(i)。该方法相对于现有技术提供了巨大的优势,这涉及可扩展性和工业实施。与公布的数据相比,总收率显著更高;此外,通常获得非常高纯度的活性物质。该新方法的一个实施方案能够可再现、经济地制备现有技术中之前未描述的特定晶形。通过本文提出的本发明方法,已经成功地制造了几千克用于临床试验的材料。
在本发明的上下文中优选的盐是本发明化合物的生理学上可接受的盐(例如参见s.m.berge等,"pharmaceuticalsalts",j.pharm.sci.1977,66,1-19)。然而,还包括本身不适用于制药应用但可例如用于本发明化合物的分离或提纯的盐。
本发明化合物的生理学上可接受的盐包括无机酸、羧酸和磺酸的酸加成盐,例如盐酸、氢溴酸、硫酸、磷酸、甲磺酸、乙磺酸、甲苯磺酸、苯磺酸、萘二磺酸、乙酸、三氟乙酸、丙酸、乳酸、酒石酸、苹果酸、柠檬酸、富马酸、马来酸和苯甲酸的盐。
本发明化合物的生理学上可接受的盐还包括常规碱的盐,优选的实例是碱金属盐(例如钠盐和钾盐)、碱土金属盐(例如钙盐和镁盐)和衍生自氨或具有1-16个碳原子的有机胺的铵盐,该有机胺的优选的实例是乙胺、二乙胺、三乙胺、乙基二异丙基胺、单乙醇胺、二乙醇胺、三乙醇胺、二环己基胺、二甲基氨基乙醇、普鲁卡因、二苄胺、n-甲基吗啉、精氨酸、赖氨酸、亚乙基二胺、n-甲基哌啶和胆碱。
在本发明的上下文中,溶剂合物被描述为以固态或液态通过与溶剂分子配位而形成络合物的本发明化合物的那些形式。水合物是溶剂合物的特定形式,其中与水配位。
式(i-a)、(xii-a)、(xiv-a)和(i-b)、(xii-b)、(xiv-b)的化合物各自是式(i)、(xii)和(xiv)的化合物的子集,并且对于1,2,4-三唑环或其受保护形式的第5位上的醇基的立体中心而言各自是对映异构体或非对映异构体。式(i-a)、(xii-a)和(xiv-a)的化合物在此被指定为立体中心的(s)构型,并且式(i-b)、(xii-b)和(xiv-a)的化合物各自被指定为立体中心的(r)构型。
如果本发明的化合物可以以互变异构形式存在,则本发明包括所有互变异构形式。
三唑衍生物的以下三个互变异构体代表(a)、(b)和(c)彼此等价且同义,并且在所有情况下都描述为1,4-二取代的三唑衍生物。
这特别适用于以下结构单元:1h-1,2,4-三唑-3-基、1h-1,2,4-三唑-5-基、4h-1,2,4-三唑-3-基和4h-1,2,4-三唑-5-基。在此,y1和y2是不同的取代基。
本发明提供了制备通式(i)的化合物或其盐、其溶剂合物或其盐的溶剂合物的方法,其特征在于,其包括步骤[c]和[d],其中
[c]使通式(xi)的化合物
其中
r2是(c1-c4)-烷基,优选甲基,
以相继的方式在第一步骤中
[c-1]在碱的存在下与通式(xii)的酰氯反应
其中
pg是保护基,优选乙酰基,
然后将所得中间产物在随后步骤中
[c-2]在碱的存在下与通式(xiii)的苯基肼化合物反应
其中r1a和r1b彼此独立地选自氢、氟、氯、甲基、单氟甲基、二氟甲基、三氟甲基、乙基、甲氧基、二氟甲氧基和三氟甲氧基,
以产生通式(xiv)的1,2,4-三唑基化合物
其中pg、r1a和r1b具有上面给出的定义,
并且将其在随后步骤中
[d]通过脱除保护基pg而转化成通式(i)的化合物
其中r1a和r1b具有上面给出的定义。
与现有技术(wo2016/071212)相反,(i)的制备(经由(xi)+(xii)+(xiii)→(xiv)→(i),见图式3:步骤3+4:通过2个阶段为36.7%至82.2%)比现有技术中的类似序列具有明显更高的收率(参见图式2:(v)→(vii)→(i),通过2个阶段为2.9%至28.9%)。
在一个有利的实施方案中,本发明的方法在与化学机理匹配的多阶段操作模式中作为一锅反应进行。
在这一上下文中,该方法在合适的溶剂存在下进行,然后使获自步骤[c-1]的中间产物不经分离,即在溶液中在随后的步骤[c-2]中转化。
在另一个有利的实施方案中,获自步骤[c-2]的通式(xiv)的1,2,4-三唑基化合物不经分离,即在溶液中在随后的步骤[d]中转化成式(i)的化合物。
在一个实施方案中,本发明的方法在步骤[c]之前包括另一步骤[b],其中
[b]使通式(x)的化合物
与碱性(c1-c4)-烷氧基化物,优选甲醇钠反应以产生通式(xi)的式的亚氨酸酯化合物
其中
r2是(c1-c4)-烷基,优选甲基。
在一个有利的实施方案中,本发明的方法在与化学机理匹配的多阶段操作模式中作为一锅反应进行。
在这一上下文中,所述反应在合适的溶剂存在下进行,并且获自步骤[b]的通式(xi)的式的亚氨酸酯化合物不经分离,即在溶液中然后在随后的步骤[c]中转化。
在一个实施方案中,本发明的方法在步骤[c]之前包括步骤[b],并且在步骤[b]之前包括另一步骤[a],其中
[a]使通式(ii)的化合物
与腈化合物(ix)反应
其中x是离去基,优选氯离子或溴离子,
以产生通式(x)的化合物
在本发明方法的另一个实施方案中,该方法包括步骤[a]、[b]、[c]和[d],其中
[a]使通式(ii)的化合物
与腈化合物(ix)反应
其中x是离去基,优选氯离子或溴离子,
以产生通式(x)的化合物
并且将其在随后的步骤中
[b]与碱性(c1-c4)-烷氧基化物,优选甲醇钠反应以产生通式(xi)的亚氨酸酯化合物,
其中
r2是(c1-c4)-烷基,优选甲基,
并且将其在随后的步骤中
[c]以相继的方式在第一步骤中
[c-1]在碱的存在下与通式(xii)的酰氯反应
其中
pg是保护基,优选乙酰基,
然后将所得中间产物在随后步骤中
[c-2]在碱的存在下与通式(xiii)的苯基肼化合物反应
其中r1a和r1b彼此独立地选自氢、氟、氯、甲基、单氟甲基、二氟甲基、三氟甲基、乙基、甲氧基、二氟甲氧基和三氟甲氧基,
以产生通式(xiv)的1,2,4-三唑基化合物,
其中r1a和r1b具有上面给出的定义,且
pg是保护基,优选乙酰基,
并且将其在随后的步骤中
[d]通过脱除保护基pg而转化成通式(i)的化合物
其中r1a和r1b具有上面给出的定义。
优选制备式(i)的化合物的方法,其特征在于,r1a和r1b彼此独立地选自氢、氟和氯,其中所述取代基的至少一个不是氢。
特别优选制备式(i)的化合物的方法,其特征在于,r1a是氢,且r1b在2位或3位是氯。
非常特别优选制备式(i)的化合物的方法,其特征在于,r1a是氢,且r1b在3位是氯。
非常特别优选制备式(i-a-1)的化合物的方法
本发明还提供了制备通式(x)的化合物或其盐、其溶剂合物或其盐的溶剂合物的方法,其特征在于,其包括步骤[a],其中
[a]使通式(ii)的化合物
与腈化合物(ix)反应
其中x是离去基,优选氯离子或溴离子,
以产生通式(x)的化合物
本发明还提供了制备通式(xi)的化合物或其盐、其溶剂合物或其盐的溶剂合物的方法,其特征在于,其包括步骤[b],其中
[b]使通式(x)的化合物
与碱性(c1-c4)-烷氧基化物,优选甲醇钠反应以产生通式(xi)的式的亚氨酸酯化合物,
其中
r2是(c1-c4)-烷基,优选甲基。
本发明还提供了通式(x)的化合物
在本发明的一个优选的实施方案中,该化合物是{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙腈(x-a)
本发明还提供了通式(x)的化合物用于制备通式(i)的化合物的方法。
本发明还提供了通式(xi)的化合物
其中
r2是(c1-c4)-烷基,优选甲基。
在本发明的一个优选的实施方案中,该化合物是2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙亚氨酸甲酯(xi-a)
本发明还提供了通式(xi)的化合物用于制备通式(i)的化合物的用途。
本发明还提供了通式(xiv)的化合物用于制备通式(i)的化合物的用途。
步骤1:适用于方法步骤[a]:(ii)+(ix)→(x)的碱是常规的无机或有机碱,例如和优选碱金属碳酸盐例如碳酸钠、碳酸钾或碳酸铯,碱金属醇盐例如叔丁醇钠或叔丁醇钾,或有机胺例如n,n-二异丙基乙基胺(dipea)和三乙胺。所用的溶剂可以是惰性溶剂,例如乙腈、甲基异丁基酮、二氧杂环己烷、二甲基甲酰胺、二甲基乙酰胺、n-甲基吡咯烷酮、二甲亚砜或环丁砜。优选使用甲基异丁基酮或乙腈中的碳酸钾。
任选地,这些方法步骤可以有利地通过添加烷基化催化剂,例如溴化锂、碘化钠、四正丁基溴化铵或苄基三乙基氯化铵来进行。此外,可以发现经较长时间缓慢地计量加入烷基化剂氯乙腈或溴乙腈是有利的。该反应通常在+40℃至+120℃,优选在+60℃至+80℃的温度范围内进行。
该反应可在标准、升高或降低的压力(例如0.5至5巴)下进行;通常在标准压力下操作。
或者,式(x)的化合物也可以由文献中已知的式(xx)的化合物制备(参见图式5):
图式5:
[pvcl=特戊酰氯,tfaa=三氟乙酸酐]。
偶联反应(xx)→(xxi)[酰胺形成]可以通过在碱存在下的缩合剂或活化剂的直接途径或通过可获自(xx)的羧酰氯、羧酸酯或羧酰基咪唑的中间体进行。
合适的这类缩合剂或活化剂是例如碳二亚胺例如n,n'-二乙基-、n,n'-二丙基-、n,n'-二异丙基-、n,n'-二环己基碳二亚胺(dcc)或n-(3-二甲基氨基丙基)-n'-乙基碳二亚胺盐酸盐(edc),光气衍生物例如n,n'-羰基二咪唑(cdi)、氯甲酸异丙基酯或氯甲酸异丁基酯、1,2-噁唑鎓化合物例如2-乙基-5-苯基-1,2-噁唑鎓-3-硫酸盐或2-叔丁基-5-甲基异噁唑鎓高氯酸盐,酰基氨基化合物例如2-乙氧基-1-乙氧基羰基-1,2-二氢喹啉、α-氯烯胺例如1-氯-n,n,2-三甲基丙-1-烯-1-胺,1,3,5-三嗪衍生物例如4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉氯化物,磷化合物例如正丙烷膦酸酐(t3p,ppaca)、氰基膦酸二乙基酯、二苯基磷酰基叠氮化物(dppa)、双(2-氧代-3-噁唑烷基)磷酰基氯、苯并三唑-1-基氧基三(二甲基氨基)鏻六氟磷酸盐或苯并三唑-1-基氧基三(吡咯烷基)鏻六氟磷酸盐(pybop)或脲鎓化合物例如o-(苯并三唑-1-基)-n,n,n',n'-四甲基脲鎓四氟硼酸盐(tbtu)、o-(苯并三唑-1-基)-n,n,n',n'-四甲基脲鎓六氟磷酸盐(hbtu)、o-(1h-6-氯苯并三唑-1-基)-1,1,3,3-四甲基脲鎓四氟硼酸盐(tctu)、o-(7-氮杂苯并三唑-1-基)-n,n,n',n'-四甲基脲鎓六氟磷酸盐(hatu)或2-(2-氧代-1-(2h)-吡啶基)-1,1,3,3-四甲基脲鎓四氟硼酸盐(tptu),其任选与其它助剂例如1-羟基苯并三唑(hobt)或n-羟基琥珀酰亚胺(hosu)组合,以及合适的碱是碱金属碳酸盐,例如碳酸钠或碳酸钾,或叔胺碱,例如三乙胺、n-甲基吗啉(nmm)、n-甲基哌啶(nmp)、n,n-二异丙基乙基胺(dipea)、吡啶或4-n,n-二甲基氨基吡啶(dmap)。通常,所述酰氯通过与亚硫酰氯或草酰氯在惰性溶剂例如二氯甲烷或n,n-二甲基甲酰胺中反应而制备。也可以使用所列溶剂的混合物。
转化成腈(xxi)→(x)可在脱水剂存在下进行。常见脱水剂是例如三氟乙酸酐(tfaa)、五氧化二磷(p4o10)、磷酰氯(pocl3)、五氯化磷(pcl5)、ccl4-pph3(appel试剂)、六甲基磷酰胺(hmpa);n-(三乙基铵磺酰基)氨基甲酸甲酯(burgess试剂)、(氯亚甲基)二甲基亚胺氯化物(vilsmeier试剂)、草酰氯/dmso和亚硫酰氯(socl2)。
用于两个方法步骤(xx)→(xxi)和(xxi)→(x)的常见溶剂例如是醚例如二乙醚、二氧杂环己烷、四氢呋喃、乙二醇二甲醚或二乙二醇二甲醚,烃例如苯、甲苯、二甲苯、己烷、环己烷或石油级分,卤代烃例如二氯甲烷、三氯甲烷、四氯甲烷、1,2-二氯乙烷、三氯乙烯或氯苯,或其它溶剂例如丙酮、乙酸乙酯、乙腈、二甲亚砜、n,n-二甲基甲酰胺、n,n'-二甲基亚丙基脲(dmpu)、n-甲基吡咯烷酮(nmp)或吡啶。可以使用所述溶剂的混合物。
通常优选地,羧酸(xx)在第一步骤中在吡啶存在下与特戊酰氯反应,由此产生中间体,其在随后的步骤中与氨反应。通常,形成的中间体不经分离,并且该反应通过两个阶段作为一锅反应进行。适用于第一步骤的碱优选是吡啶、4-(n,n-二甲基氨基)吡啶或n,n-二异丙基乙基胺(dipea)。然后,通常经由与三氟乙酸酐的反应使羧酰胺(xx)转化成腈(x)。这两个反应均在惰性有机溶剂,优选四氢呋喃中进行。
式(xx)的化合物可从文献中获知(参见wo2010/105770,图式2,实施例8a和9a;和wo2011/104322,图式11)。
步骤2:可用于在步骤[b]中制备亚氨酸酯(xi)的碱是碱性(c1-c4)-碱金属醇盐,例如甲醇钠、乙醇钠、丙醇钠、异丙醇钠、叔丁醇钠或叔丁醇钾。合适的醇是醇,例如甲醇、乙醇、正丙醇、2-丙醇、正丁醇、2-丁醇和叔丁醇。优选使用甲醇中的甲醇钠。
该反应通常在+20至+80℃,优选在+40至+60℃的温度范围内进行。亚氨酸酯(xi)不必中间分离,但可以直接通过从甲醇变为甲苯或四氢呋喃的蒸馏而用于随后阶段。
步骤3:方法步骤[c]中的多组分环化反应在两阶段法中进行。首先,在步骤[c-1]中,亚氨酸酯(xi)在碱存在下与酰氯(xii)反应,然后所得中间体与苯基肼化合物(xiii)在步骤[c-2]中在碱存在下反应。通常,形成的中间体不经分离,并且该两阶段法作为一锅反应进行。
有利地,酰氯(xii)在此在步骤[c-1]中以1.1至1.5摩尔的量,优选以1.2摩尔的量使用,基于1摩尔的式(xi)的化合物计。步骤[c-1]中的碱通常以1至2.5摩尔的量,优选以1.05至2.0摩尔的量,更优选以1.05至1.5摩尔的量使用,基于1摩尔的式(xii)的化合物计。
步骤[c-2]中的肼(xiii)也可以以盐的形式,例如作为盐酸盐或作为对-甲苯磺酸盐(甲苯磺酸盐)使用。在碱性反应条件下,此时盐形式转化成游离肼。在这种情况下,可以相应地调节碱量。在另一个有利的实施方案中,在加入之前将肼盐在单独的反应容器中中和,然后将所得溶液任选在过滤除去所形成的盐之后作为溶液加入反应混合物中。
步骤[c-2]中的碱通常以1.05至1.5摩尔的量,优选以1.2至1.5摩尔的量使用,基于在1摩尔式(xiii)的化合物计。
适用于这两个步骤的碱通常为叔胺碱,例如n,n-二异丙基乙基胺(dipea)、三乙胺、三异丙基胺、n-甲基咪唑、n-甲基吗啉、吡啶和4-(二甲基氨基)吡啶。优选三乙胺或n,n-二异丙基乙基胺。特别优选二异丙基乙基胺。
合适的溶剂是惰性有机溶剂,例如二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、四氢呋喃、1,4-二氧杂环己烷、1,2-二甲氧基乙烷、甲苯、吡啶、乙酸乙酯、乙腈或n,n-二甲基甲酰胺或这些溶剂的混合物。
优选使用四氢呋喃(thf)或四氢呋喃和甲苯的混合物。
在步骤[c-1]中与酰氯(xii)的反应和在步骤[c-2]中与肼(xiii)的反应在-20℃至+30℃,优选在0℃至+10℃的温度范围内进行。对于在步骤[c-2]中通过消去水而形成三唑(环化),随后使反应混合物达到+20至+150℃的温度。优选地,该反应在+70至+80℃的温度下进行。
步骤4:在工艺步骤[d]中保护基pg的引入和脱除是通过文献常规的方法进行的[参见例如t.w.greene和p.g.m.wuts,protectivegroupsinorganicsynthesis,wiley,newyork,1999]。例如,优选用碱,例如氢氧化钠水溶液除去乙酰基。
当方法步骤[d]中保护基脱除通过氢氧化钠水溶液进行时,后处理例如通过用合适溶剂萃取,然后重复洗涤和干燥进行。优选用甲基叔丁基醚(mtbe)萃取。
式(ii)、(ix)、(xii)、(xiii)和(xx)的化合物可商购或本身描述在文献中,或者它们可以类似于在文献中公开的方法以本领域技术人员显而易见的方式制备。用于制备起始材料的许多详细规程和文献信息存在于实验部分中。
由于式(i-a-1)的化合物以片剂的形式研发,因此对经分离的式(i-a-1)的化合物可再现性地以特定晶形分离的需求很高,以使得可以确保可再现的生物利用度。
令人惊讶地发现,式(i-a-1)的化合物(5-(4-氯苯基)-2-({1-(3-氯苯基)-5-[(1s)-1-羟基乙基]-1h-1,2,4-三唑-3-基}甲基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮
可以从甲基叔丁基醚/二异丙基醚的混合物中,或从甲基叔丁基醚/正-庚烷的混合物中结晶,其中可再现地形成结晶多形体i(步骤5)。
步骤5:在此,式(i-a-1)的化合物在5至10倍过量的甲基叔丁基醚(mtbe)中的溶液在20至80℃,优选50至60℃,更优选在甲基叔丁基醚的回流温度(约54℃)下搅拌,并在此温度下加入二异丙基醚。在连续添加二异丙基醚的情况下,蒸馏出mtbe。在此,式(i-a-1)的化合物结晶出来。冷却至0至30℃,优选10至20℃的温度,并且将晶体分离并在真空中在40至60℃,优选40至50℃下干燥。
或者,可以使用甲基叔丁基醚/正-庚烷的混合物。这里,式(i-a-1)的化合物在5至10倍过量的甲基叔丁基醚(mtbe)中的溶液在20至80℃,优选50至60℃,更优选在甲基叔丁基醚的回流温度(约54℃)下搅拌,在此温度下加入1.5至2.5倍体积的正-庚烷,由此式(i-a-1)的化合物结晶出来。冷却至0至30℃,优选10至20℃的温度,并将晶体分离并在真空中在40至80℃,优选40至50℃下干燥。
出于gmp技术的原因,可能适宜的是在加热之前首先使产物在mtbe中的溶液经历颗粒过滤。
后处理通常通过过滤、用二异丙基醚或正-庚烷重复洗涤,然后干燥来进行。
达到的>99%的化学纯度和约100%的含量符合根据ich指南的商业产品标准。光学纯度为>>99%e.e。
结晶过程非常稳健,并以可再现的方式提供所需的晶形。式(i)的化合物通常被微粉化,并且在制药中配制成片剂。已经发现该晶形具有非常好的稳定性(即使在高空气湿度下)并且可以经数月储存而不损失稳定性。
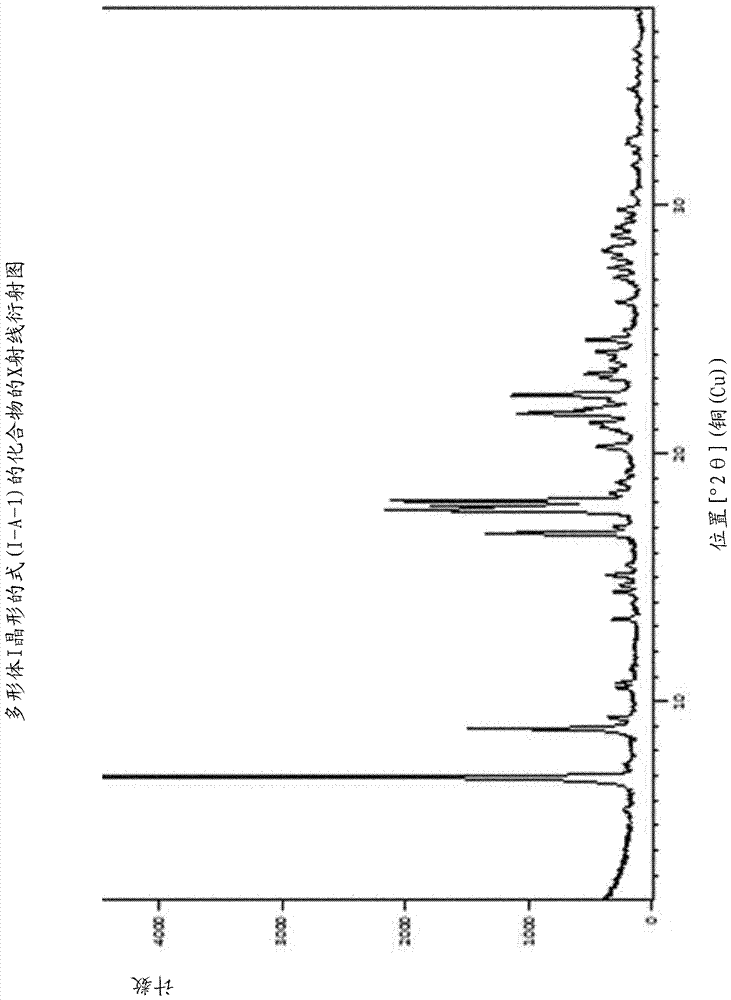
本发明还提供了多形体i晶形的式(i-a-1)的化合物(5-(4-氯苯基)-2-({1-(3-氯苯基)-5-[(1s)-1-羟基乙基]-1h-1,2,4-三唑-3-基}甲基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮。
本发明提供了多形体i晶形的式(i-a-1)的化合物,其特征在于,该化合物的x射线衍射图显示在7.0、8.9、16.8、17.7、17.9、18.1、21.6、21.8、22.4和24.6处的2θ角的最大峰值。
本发明还提供了制备多形体i晶形的式(i-a-1)的化合物的方法,其特征在于,以一种或多种多形体或以溶剂合物形式存在的式(i-a-1)的化合物在甲基叔丁基醚/二异丙基醚的混合物中或甲基丁基醚/正-庚烷的混合物中在20℃至80℃的温度下搅拌,然后过滤,洗涤并在真空中干燥。
用于制备多形体i晶形的式(i-a-1)的化合物的方法的优选溶剂是甲基叔丁基醚/二异丙基醚的混合物或甲基叔丁基醚/正-庚烷的混合物。
用于制备多形体i晶形的式(i-a-1)的化合物的方法的优选温度范围是甲基叔丁基醚的回流温度(约54℃)。
化合物还提供了用于治疗疾病的如上所述的多形体i晶形的式(i-a-1)的化合物。
本发明还提供了包含如上所述的多形体i晶形的式(i-a-1)的化合物并且没有较大含量的不同于如上所述的多形体i晶形的式(i-a-1)的化合物的其它形式的药物。本发明还提供了包含基于所包含的式(i-a-1)的化合物的总量计大于90重量%的如上所述的多形体i晶形的式(i-a-1)的化合物的药物。
本发明还提供了如上所述的多形体i晶形的式(i-a-1)的化合物用于制造治疗心血管疾病和肾病的药物的用途。
本发明还提供了通过给予有效量的如上所述的多形体i晶形的式(i-a-1)的化合物治疗心血管疾病和肾疾的方法。
本发明的式(i-a-1)的化合物充当有效的双重v1a/v2受体拮抗剂,并表现出不可预料、有价值的药理作用谱。因此,它们适合用作治疗和/或预防人类和动物疾病的药物。
本发明的化合物本身或与一种或多种其它活性物质组合着适合于预防和/或治疗各种疾病,例如心血管系统的疾病(心血管疾病),适合于心脏损伤后的心脏保护,和代谢和肾脏疾病。
本发明的化合物本身或与一种或多种其它活性物质组合着适合于预防和/或治疗各种疾病,例如心血管系统的疾病(心血管疾病)和肾病。
本发明的化合物具有有价值的药理性质并且可用于预防和/或治疗人类和动物的各种疾病和疾病相关的病情。
示例性且优选地在wo2016/071212,第16至19页中列出了可能的目标指征。
示例性且优选地在在wo2016/071212,第19至27页中列出了合适的组合活性物质和剂型。
以下实施例说明了本发明。本发明不受限于这些实施例。
除非另有说明,否则下面的试验和实施例中的百分率是重量百分率;份是重量份。液体/液体溶液的溶剂比、稀释比和浓度数据在每种情况下基于体积。
a.实施例
缩写:
aq.水性,水溶液
c浓度
cat.催化性
cdin,n'-羰基二咪唑
dci直接化学电离(在ms中)
dest.经蒸馏
diean,n-二异丙基乙基胺
dmap4-n,n-二甲基氨基吡啶
dmf二甲基甲酰胺
dmso二甲亚砜
d.th.理论值的(在收率中)
ee对映体过量
ent对映体纯,对映异构体
eq.当量
esi电喷雾电离(在收率中)
et乙基
gc-ms气相色谱-质谱联用
h小时
hplc高压、高效液相色谱
konz.浓缩
lc-ms液相色谱-质谱联用
me甲基
min分钟
ms质谱
mtbe甲基叔丁基醚
nmr核磁共振波谱
ph苯基
quant.定量(在收率中)
rac外消旋,外消旋物
rt室温
rt滞留时间(在hplc中)
thf四氢呋喃
uv紫外光谱
v/v(溶液的)体积/体积比。
lc/ms和hplc方法:
方法1(lc/ms):mcw-sq-hsst3
仪器:watersacquitysqduplcsystem;柱:watersacquityuplchsst31.8µ,50mmx1mm;洗脱剂a:1l水+0.25ml99%甲酸,洗脱剂b:1l乙腈+0.25ml99%甲酸;梯度:0.0min90%a→1.2min5%a→2.0min5%a;炉:50℃;流速:0.40ml/min;uv检测:208-400nm。
方法2(lc/ms):mcw-ft-ms-m1
仪器:thermoscientificft-ms;仪器uhplc+:thermoscientificultimate3000;柱:waters,hsst3,2.1x75mm,c181.8µm;洗脱剂a:1l水+0.01%甲酸;洗脱剂b:1l乙腈+0.01%甲酸;梯度:0.0min10%b→2.5min95%b→3.5min95%b;炉:50℃;流速:0.90ml/min;uv检测:210nm/最佳积分路径210-300nm。
进一步说明:
除非另外说明,下面实施例和测试说明中的百分率是重量百分率;份是重量份。液体/液体溶液的溶剂比、稀释比和浓度数据在每种情况下基于体积。
在通过上述方法通过制备型hplc提纯本发明化合物的情况下(其中洗脱剂含有添加剂例如三氟乙酸、甲酸或氨),本发明的化合物以盐的形式,例如作为三乙酸盐、甲酸盐或铵盐获得,如果本发明的化合物含有足够碱性或酸性的官能团。这种盐可以通过本领域技术人员已知的各种方法转化成相应的游离碱或酸。
纯度数据通常基于lc/ms色谱图中的相应峰积分,但是也还可以借助1hnmr波谱测定。如果没有说明纯度,则通常是指根据lc/ms色谱图中的自动化峰积分的100%纯度,或纯度未明确测定。
如果说明<100%的纯度,则以理论值的%为单位的所示收率通常是针对纯度经校正的。在含有溶剂或受污染的批次中,形式收率可能是“>100%”;在这些情况下,收率没有针对溶剂或纯度进行校正。
下面1hnmr信号的耦合模式的描述已部分地直接取自acdspecmanager(acd/labsrelease12.00,产品版本12.5)的建议并且不一定严格探究。在某些情况下,specmanager的建议已经手动调整。手动调整或指定的描述通常基于所涉信号的光学外观,并且不一定对应于严格、物理准确的解读。通常,所示的化学位移基于所涉信号中心。在宽多重峰的情况下,给出区间。由溶剂或水遮蔽的信号试验性赋值或未列出。严重加宽的信号-例如由分子部分的快速旋转或由于交换的质子引起-同样试验性赋值(通常称为宽多重峰或宽单峰)或未列出。
所选实施例的1hnmr数据以1hnmr峰列表的形式阐述。对于每个信号峰,首先将δ值以ppm,然后将信号强度以圆括号的形式列出。各信号峰的一对δ值/信号强度值通过逗号彼此分开地列出。因此,一个实例的峰列表具有以下形式:δ1(强度1),δ2(强度2),...,δi(强度i),...,δn(强度n)。
尖信号的强度与打印的nmr波谱实例中的以cm为单位的信号高度相关联,并显示出信号强度相比于其它信号的真实比例。在宽信号的情况下,可以显示多个峰或这些峰的中心及其相比于波谱中的最强信号而言的相对强度。1hnmr峰的列表类似于传统1hnmr打印输出,并因此通常含有列在传统nmr解读中的所有峰。此外,如传统1hnmr打印输出那样,它们可显示溶剂信号、同样通过本发明提供的目标化合物的立体异构体的信号和/或杂质信号。目标化合物的立体异构体的峰和/或杂质的峰通常具有比目标化合物(例如纯度>90%)的峰平均更低的强度。这种立体异构体和/或杂质可以对于各制备方法而言是典型的。因此,它们的峰可以有助于借助“副产物指纹”鉴定我们制备方法的再现性。如果需要,通过已知方法(mestrec,acd模拟或使用经验评估的预期值)计算目标化合物的峰值的专家可以分离目标化合物的峰,其中任选地使用额外的强度滤波器。这一分离将与传统1hnmr解读中的所涉的峰拣选相似。在出版物“citationofnmrpeaklistdatawithinpatentapplications”中可获知峰列表形式的nmr数据阐述的详细描述(参见researchdisclosuredatabasenumber605005,2014,2014年8月1日或http://www.researchdisclosure.com/searching-disclosures)。在researchdisclosuredatabasenumber605005中描述的峰拣选程序中,参数“minimumheight”可以设定为1%至4%。取决于化学结构的类型和/或取决于要测量的化合物的浓度,可能适宜的是将参数“minimumheight”设定为<1%的值。
如果示出,熔点和熔程是未校正的。
对于下文中未明确描述其制备的所有反应物或试剂而言适用的是,它们从一般可获得的来源购买。对于其制备同样未在下文中描述且不能商业获得或从通常不可获得的来源获得的所有其它反应物或试剂,参考其中描述了其制备的公开文献。
实施例
实施例1
{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙腈(x-a)
最初装载作为在1.0l甲基异丁基酮中的溶液的100g(0.325mol)的5-(4-氯苯基)-4-((2s)-3,3,3-三氟-2-羟基丙基)-2,4-二氢-3h-1,2,4-三唑-3-酮(ii-a)(合成如wo2010/105770a1的实施例5a所述)与135g(0.975mol)碳酸钠,然后将混合物加热到60℃。随后在该温度下,经6h的时间均匀地逐滴添加溶于270ml甲基异丁基酮中的27g(0.358mol)氯乙腈(ix)。在60℃下继续搅拌15h,然后冷却到20℃,添加500ml水,继续搅拌,分离出有机相。将该有机相用500ml水再次洗涤,然后在真空中在60℃的夹套温度下浓缩到约250ml的体积。随后,添加250ml正-庚烷,由此产物结晶出来。为了完成结晶,通过同时添加500ml正-庚烷在60℃的夹套温度下在真空中蒸馏出约500ml溶剂混合物。冷却到20℃并在这一温度下搅拌一小时。过滤出产物并用正-庚烷(2x150ml)后洗涤。在40℃下在真空中干燥。收率:81g固体(理论值的72%)。
ms(eipos):m/z=347.1[m+h]+
1h-nmr(400mhz,dmso-d6):δ=3.81(dd,1h),3.98(dd,1h),4.23-4.34(m,1h),5.17(s,2h),6.91(d,1h),7.55(d,2h),7.78(d,2h)。
实施例2
2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙亚氨酸甲酯(xi-a)
最初装载作为1.6l甲醇中的溶液的200g(576.9mmol){3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙腈(x-a),添加5.2g(28mmol)甲醇钠(在甲醇中30%)。在50℃下搅拌2小时,然后在50℃的夹套温度下浓缩以产生油性残余物。添加2lmtbe,浓缩到约0.8l的体积。然后将这一溶液在搅拌下缓慢地计量加入到4l正-己烷中。在这一过程中,产物作为稠的晶体悬浮体结晶出来。冷却到20℃并在室温下搅拌一小时。过滤出产物并用正-己烷(2x0.25l)洗涤。产物在40℃下在真空中干燥。收率:175g固体(理论值的80%)。
1h-nmr(400mhz,dmso-d6):δ=3.67(s,3h),3.81(dd,1h),3.96(dd,1h),4.23-4.35(m,1h),4.50(s,2h),6.93(br.s,1h),7.62(d,2h),7.78(d,2h),8.01(s,1h)。
实施例3
(5-(4-氯苯基)-2-({1-(3-氯苯基)-5-[(1s)-1-羟基乙基]-1h-1,2,4-三唑-3-基}甲基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(i-a-1)
方法变体b:
将2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙亚氨酸甲酯(xi-a)(164g,433mmol)溶解在thf(1.0l)和甲苯(0.5l)的混合物中。向该混合物加入n-乙基二异丙基胺(97.8g,757mmol),然后在20℃下继续搅拌15min。随后在0℃下计量加入(s)-2-乙酰氧基丙酰氯(xii-a)(78.2g,519mmol),在0℃下继续搅拌1h。随后在0℃下计量加入4-氯苯基肼盐酸盐(xiii-1)(85.2g,476mmol)和n-乙基二异丙基胺(67.11g,519mmol)在thf(0.5l)中的溶液,其中在计量加入前过滤除去沉淀的n-乙基二异丙基胺盐酸盐,然后在20℃下继续搅拌1h和在回流温度(约75℃)下另外2h。冷却到20℃,将0.75l水添加到该混合物中。相分离后,有机相用每次0.5l1n盐酸溶液洗涤两次,然后在真空中在80℃的夹套温度下浓缩成油性残余物,并用每次1.0l甲醇共蒸馏两次。然后将油性残余物溶于0.6l甲醇中,在0℃添加0.5l1n氢氧化钠溶液,并在20℃下继续搅拌1小时。添加0.75l水和0.75lmtbe后,分离出有机相,用每次0.3l半饱和氯化钠水溶液洗涤两次,然后在80℃的夹套温度下在真空中浓缩直至约0.3l的体积。添加1.5l二异丙基醚后,再次在80℃的夹套温度下在真空中浓缩直至约0.3l的体积,由此产物沉淀出来。冷却到10℃并在该温度下搅拌1小时。过滤出产物并用0.3l二异丙基醚后洗涤。在50℃下在真空中干燥。收率:200g(理论值的72%)。
ms(esipos):m/z(%)=543.1(100)[m+h]+.
1h-nmr(400mhz,dmso-d6):δ=1.47(d,3h),3.85(dd,1h),4.00(dd,1h),4.24-4.36(m,1h),4.81(quin,1h),5.07(s,2h),5.75(d,1h),6.89(d,1h),7.54-7.66(m,5h),7.72-7.79(m,3h)。
方法变体c(通过随后从甲基叔丁基醚/二异丙基醚中结晶):
最初装载作为6.9l甲醇中的溶液的1.373kg(3.96mol){3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙腈(x-a),添加36g(0.198mol)甲醇钠(在甲醇中30%)。在50℃下搅拌1.5小时,然后在50℃的夹套温度下浓缩以产生仍可搅拌的浆状残余物。添加各3.0l甲苯三次,并在每种情况下浓缩至5l的体积。将thf(9.5l)和甲苯(2.5l)添加到残余物中,在20℃下添加n-乙基二异丙基胺(0.896kg,6.93mol),并在20℃下继续搅拌15分钟。随后在0℃下,计量加入(s)-2-乙酰氧基丙酰氯(xii-a)(0.715kg,4.752mol),并在0℃下继续搅拌1小时。
随后,在0℃下计量加入4-氯苯基肼盐酸盐(xiii-1)(0.78kg,4.356mol)和n-乙基二异丙基胺(0.614kg,4.752mol)在thf(4.5l)中的溶液,其中在计量加入之前过滤除去沉淀的n-乙基二异丙基胺盐酸盐,然后在20℃搅拌1小时并在回流温度(约75℃)下继续搅拌另外2小时。冷却到20℃,并将7.0l水添加到该混合物中。相分离后,将有机相用每次3.5l1n盐酸溶液洗涤两次,并在80℃的夹套温度下在真空中浓缩成油性残余物,并与每次13.5l甲醇共蒸馏两次。
将该油性残余物溶于5.5l甲醇中,在0℃下添加4.0l1n氢氧化钠溶液,并在20℃下继续搅拌1小时。添加7.0l水和7.0lmtbe后,分离出有机相,用每次2.75l半饱和氯化钠水溶液洗涤两次,并在80℃的夹套温度下在真空中浓缩直至约3.0l的体积。添加16.0l二异丙基醚后,再次在80℃的夹套温度下在真空中浓缩直至约6.0l的体积,由此产物沉淀出来。然后冷却到10℃并在该温度下搅拌1小时。过滤出产物,并用每次1.0l二异丙基醚后洗涤两次。在50℃下在真空中干燥。收率:1.680kg(理论值的78%)。
>99%的纯度;>>99%e.e的光学纯度。
方法变体c(通过随后从甲基叔丁基醚/正-庚烷中结晶):
最初装载作为250ml甲醇中的溶液的50g(144mmol)3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙腈(x-a),添加1.58g(7.3mmol)甲醇钠(在甲醇中25%)。在50℃下搅拌1.5小时,然后在50℃的夹套温度下浓缩以产生仍可搅拌的浆状残余物。添加每次200mldmf三次,并在真空中浓缩至干。将325mlthf添加到残余物中,在20℃下添加n-乙基二异丙基胺(44ml,253mmol),并在20℃下继续搅拌15分钟。随后在0℃下,计量加入(s)-2-乙酰氧基丙酰氯(xii-a)(26g,173mmol),在0℃下继续搅拌1小时。随后,在0℃下计量加入4-氯苯基肼盐酸盐(xiii-1)(28.5g,159mmol)和n-乙基二异丙基胺(30ml,172mmol)在150mlthf中的溶液,其中在计量加入之前过滤除去沉淀的n-乙基二异丙基胺盐酸盐。随后,在20℃下继续搅拌30分钟和在回流温度(约75℃)下另外2.5小时。冷却到20℃,并将125mlmtbe和250ml水添加到该混合物中。相分离后,将有机相用每次125g1n盐酸溶液洗涤两次,在60℃的夹套温度下在真空中浓缩成油性残余物,并与每次500ml甲醇共蒸馏两次。将该油性残余物溶于200ml甲醇中,在0℃下添加35ml1n氢氧化钠溶液,并在20℃下继续搅拌1小时。添加175ml水和375mlmtbe后,除去有机相,用每次62ml半饱和氯化钠水溶液洗涤两次,并在80℃的夹套温度下在真空中浓缩成油性残余物。添加300ml二异丙基醚后,再次在80℃的夹套温度下在真空中浓缩直至约150ml的体积,由此产物沉淀出来。然后冷却到10℃并在该温度下搅拌1小时。过滤出产物,并用每次100ml二异丙基醚洗涤两次。在50℃下在真空中干燥。在回流下将晶体溶于420mlmtbe中。在50℃下添加900ml正-庚烷后,产物结晶出来。然后冷却到20℃并在该温度下搅拌1小时。过滤出产物,用100ml正-庚烷后洗涤,并在70℃下在真空中干燥。收率:58g(理论值的74%)。
>99%的纯度;光学纯度>>99%e.e。
实施例4
a)乙酸(1r)-1-[1-(3-氯苯基)-3-({3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}甲基)-1h-1,2,4-三唑-5-基]乙基酯(xiv-b-1)
在用冰冷却的同时,将87mg(0.58mmol)的(r)-(-)-2-乙酰氧基丙酰氯(xii-b)逐滴添加到200mg(0.53mmol)的2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙亚氨酸甲酯(xi)和262µl(1.5mmol)dipea在2mlthf中的混合物中。在0℃下1小时后,添加104mg(0.58mmol)的3-氯苯基肼(xiii),然后将该混合物在室温下搅拌过夜。通过色谱法(制备型hplc,洗脱剂:乙腈/水梯度,0.1%甲酸)提纯反应混合物。将含产物的级分冷冻干燥后,产生208mg(理论值的64%)标题化合物。
lc-ms(方法2):rt=2.04min;ms(esipos):m/z=585.1[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.90-7.37(m,8h),6.89(d,1h),5.91(d,1h),5.09(s,2h),4.40-4.20(m,1h),4.09-3.71(m,2h),1.81(s,3h),1.56(d,3h)。
b)2-({1-(3-氯苯基)-5-[(1r)-1-羟基乙基]-1h-1,2,4-三唑-3-基}甲基)-5-(4-氯苯基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(i-b-1)
将200mg(0.34mmol)来自步骤a)的化合物和341µl(0.34mmol)1m氢氧化钠溶液在2.6ml甲醇中的混合物在室温下搅拌30分钟。添加1g活化的离子交换剂(dowex50wx8,200-400目),在室温下搅拌5分钟。然后过滤出离子交换剂,并用甲醇后洗涤。蒸发浓缩滤液。这产生168mg(理论值的90%)标题化合物。
lc-ms(方法2):rt=1.85min;ms(esipos):m/z=543.1[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.98-7.48(m,8h),6.90(d,1h),5.76(d,1h),5.07(s,2h),4.81(t,1h),4.46-3.68(m,3h),1.47(d,3h)。
实施例5
a)乙酸(1s)-1-[3-({3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}甲基)-1-(2,4-二氯苯基)-1h-1,2,4-三唑-5-基]乙基酯(xiv-a-2)
在用冰冷却的同时,将73µl(0.58mmol)的(s)-(-)-2-乙酰氧基丙酰氯(xii-a)逐滴添加到200mg(0.53mmol)的2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙亚氨酸甲酯(xi)和262µl(1.51mmol)dipea在2mlthf中的混合物中。在0℃下1小时后,添加124mg(0.58mmol)的2,4-二氯苯基肼盐酸盐(xiii),然后将该混合物在室温下搅拌过夜。将反应混合物在回流下加热2小时,并在微波中在100℃下加热5小时。在真空中除去溶剂,粗产物经色谱(制备型hplc,洗脱剂:乙腈/水梯度,0.1%甲酸)提纯。将含产物的级分冷冻干燥后,产生163mg(理论值的48%)标题化合物。
lc-ms(方法a):rt=1.13min;ms(esipos):m/z=619.0[m+h]+
1hnmr(dmso-d6,400mhz):δ=8.04-7.49(m,7h),6.89(d,1h),5.90-5.44(m,1h),5.10(d,2h),4.45-4.16(m,1h),4.11-3.73(m,2h),1.81(s,3h),1.53(d,3h)。
b)5-(4-氯苯基)-2-({1-(2,4-二氯苯基)-5-[(1s)-1-羟基乙基]-1h-1,2,4-三唑-3-基}甲基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(i-a-2)
将160mg(0.26mmol)来自步骤a)的化合物和258µl(0.26mmol)1m氢氧化钠溶液在2ml甲醇中的混合物在0℃下搅拌2分钟和在室温下90分钟。添加1g活化的离子交换剂(dowex50wx8,200-400目),在室温下搅拌30分钟。然后过滤出离子交换剂,并用甲醇后洗涤。蒸发浓缩滤液,将残余物在真空中干燥。这产生148mg(定量)标题化合物。
lc-ms(方法a):rt=1.02min;ms(esipos):m/z=577.1[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.92(d,1h),7.78-7.71(m,2h),7.68-7.58(m,4h),6.89(d,1h),5.52(d,1h),5.06(d,2h),4.64(s,1h),4.43-4.21(m,1h),4.08-3.72(m,2h),1.39(d,3h)。
实施例6
a)乙酸(1s)-1-{3-({3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}甲基)-1-[2-(二氟甲氧基)苯基]-1h-1,2,4-三唑-5-基}乙基酯(xiv-a-3)
在用冰冷却的同时,将55µl(0.44mmol)的(s)-(-)-2-乙酰氧基丙酰氯(xii-a)逐滴添加到150mg(0.40mmol)的2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙亚氨酸甲酯(xi)和207µl(1.19mmol)dipea在1.5mlthf中的混合物中。在0℃下30分钟后,添加76mg(0.44mmol)的2-二氟甲氧基苯基肼(xiii),然后将该混合物在室温下搅拌过夜。然后将反应混合物在微波中在100℃下加热3小时。将几滴水添加到反应混合物中,经色谱(制备型hplc,洗脱剂:乙腈/水梯度,0.1%甲酸)提纯。将含产物的级分冷冻干燥后,产生142mg(理论值的58%)标题化合物。
lc-ms(方法a):rt=1.09min;ms(esipos):m/z=617.1[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.83-6.82(m,10h),5.69(d,1h),5.09(d,2h),4.30(d,1h),4.07-3.77(m,2h),1.77(s,3h),1.53(d,3h)。
b)5-(4-氯苯基)-2-({1-[2-(二氟甲氧基)苯基]-5-[(1s)-1-羟基乙基]-1h-1,2,4-三唑-3-基}甲基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(i-a-3)
将132mg(0.21mmol)来自步骤a)的化合物和214µl(0.21mmol)1m氢氧化钠溶液在1.3ml甲醇中的混合物在0℃下搅拌2分钟和在室温下90分钟。添加0.5g活化的离子交换剂(dowex50wx8,200-400目),在室温下搅拌30分钟。然后过滤出离子交换剂,并用甲醇后洗涤。蒸发浓缩滤液,将残余物在真空中干燥。这产生117mg(95%理论值的)的标题化合物。
lc-ms(方法a):rt=0.99min;ms(esipos):m/z=575.3[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.75(d,2h),7.67-7.54(m,4h),7.45-6.97(m,3h),6.89(d,1h),5.48(d,1h),5.19-4.94(m,2h),4.61(quin,1h),4.30(d,1h),4.09-3.76(m,2h),1.39(d,3h)。
实施例7
a)乙酸(1r)-1-{3-({3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}甲基)-1-[2-(二氟甲氧基)苯基]-1h-1,2,4-三唑-5-基}乙基酯(xiv-b-3)
在用冰冷却的同时,将73µl(0.58mmol)的(r)-(-)-2-乙酰氧基丙酰氯(xii-b)逐滴添加到200mg(0.53mmol)的2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙亚氨酸甲酯(xi)和276µl(1.58mmol)dipea在2mlthf中的混合物中。在0℃下30分钟后,添加101mg(0.58mmol)的2-二氟甲氧基苯基肼(xiii),然后将该混合物在室温下搅拌过夜。然后将反应混合物在微波中在150℃下加热3小时。将几滴水添加到反应混合物中,经色谱(制备型hplc,洗脱剂:乙腈/水梯度,0.1%甲酸)提纯。将含产物的级分冷冻干燥后,产生202mg(理论值的62%)标题化合物。
lc-ms(方法a):rt=1.09min;ms(esipos):m/z=617.3[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.89-6.81(m,10h),5.79-5.59(m,1h),5.09(d,2h),4.35-4.22(m,1h),4.09-3.78(m,2h),1.76(s,3h),1.53(d,3h)。
b)5-(4-氯苯基)-2-({1-[2-(二氟甲氧基)苯基]-5-[(1r)-1-羟基乙基]-1h-1,2,4-三唑-3-基}甲基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(i-b-3)
将192mg(0.31mmol)来自步骤a)的化合物和310µl(0.31mmol)1m氢氧化钠溶液在1.9ml甲醇中的混合物在0℃下搅拌2分钟和在室温下90分钟。添加0.5g活化的离子交换剂(dowex50wx8,200-400目),在室温下搅拌30分钟。然后过滤出离子交换剂,并用甲醇后洗涤。蒸发浓缩滤液,将残余物在真空中干燥。这产生172mg(理论值的96%)的标题化合物。
lc-ms(方法a):rt=0.99min;ms(esipos):m/z=575.3[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.75(d,2h),7.68-7.53(m,4h),7.46-6.96(m,3h),6.91(d,1h),5.48(d,1h),5.06(s,2h),4.61(t,1h),4.30(d,1h),4.07-3.75(m,1h),1.39(d,1h)。
实施例8
a)乙酸(1s)-1-{3-({3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}甲基)-1-[2-氯-4-(三氟甲基)苯基]-1h-1,2,4-三唑-5-基}乙基酯(xiv-a-4)
在用冰冷却的同时,将55µl(0.44mmol)的(s)-(-)-2-乙酰氧基丙酰氯(xii-a)逐滴添加到150mg(0.40mmol)的2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙亚氨酸甲酯(xi)和207µl(1.19mmol)dipea在1.5mlthf中的混合物中。在0℃下30分钟后,添加91mg(0.44mmol)的2-氯-4-(三氟甲基)苯基肼(xiii),在室温下搅拌1小时。然后将反应混合物在微波中在100℃下加热3小时。将几滴水添加到反应混合物中,经色谱(制备型hplc,洗脱剂:乙腈/水梯度,0.1%甲酸)提纯。将含产物的级分冷冻干燥后,产生111mg(理论值的43%)标题化合物。
lc-ms(方法a):rt=1.21min;ms(esipos):m/z=653.1[m+h]+
1hnmr(dmso-d6,400mhz):δ=8.25(s,1h),8.06-7.87(m,2h),7.81-7.54(m,4h),6.89(d,1h),5.75(s,1h),5.12(d,2h),4.29(d,1h),4.07-3.77(m,2h),1.76(s,3h),1.55(d,3h)。
b)5-(4-氯苯基)-2-({1-[2-氯-4-(三氟甲基)苯基]-5-[(1s)-1-羟基乙基]-1h-1,2,4-三唑-3-基}甲基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(i-a-4)
将104mg(0.16mmol)来自步骤a)的化合物和160µl(0.16mmol)1m氢氧化钠溶液在1ml甲醇中的混合物在0℃下搅拌2分钟和在室温下90分钟。添加0.5g活化的离子交换剂(dowex50wx8,200-400目),在室温下搅拌30分钟。然后过滤出离子交换剂,并用甲醇后洗涤。蒸发浓缩滤液,将残余物在真空中干燥。这产生94mg(理论值的96%)的标题化合物。
lc-ms(方法a):rt=1.06min;ms(esipos):m/z=611.1[m+h]+
1hnmr(dmso-d6,400mhz):δ=8.18(s,1h),7.99-7.82(m,2h),7.75(d,2h),7.62(d,2h),6.89(d,1h),5.54(d,1h),5.08(d,2h),4.71(t,1h),4.29(d,1h),4.11-3.77(m,2h),1.41(d,3h)。
实施例9
a)乙酸(1s)-1-[1-(2-氯-6-氟苯基)-3-({3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}甲基)-1h-1,2,4-三唑-5-基]乙基酯(xiv-a-5)
在用冰冷却的同时,将55µl(0.44mmol)的(s)-(-)-2-乙酰氧基丙酰氯(xii-a)逐滴添加到150mg(0.40mmol)的2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙亚氨酸甲酯(xi)和207µl(1.19mmol)dipea在1.5mlthf中的混合物中。在0℃下30分钟后,添加70mg(0.44mmol)的2-氯-6-氟苯基苯基肼(xiii),在室温下搅拌1小时。然后将反应混合物在微波中在100℃下加热3小时。将几滴水添加到反应混合物中,经色谱(制备型hplc,洗脱剂:乙腈/水梯度,0.1%甲酸)提纯。将含产物的级分冷冻干燥后,产生139mg(理论值的58%)的作为阻转异构体混合物的标题化合物。
lc-ms(方法a):rt=1.10min;ms(esipos):m/z=603.1[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.82-7.50(m,7h),6.89(d,1h),5.73(d,1h),5.25-5.04(m,2h),4.43-4.19(m,1h),4.10-3.78(m,2h),1.79(s,3h),1.54(m,3h)。
b)2-({1-(2-氯-6-氟苯基)-5-[(1s)-1-羟基乙基]-1h-1,2,4-三唑-3-基}甲基)-5-(4-氯苯基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(i-a-5)
将129mg(0.21mmol)来自步骤a)的化合物和214µl(0.21mmol)1m氢氧化钠溶液在1.3ml甲醇中的混合物在0℃下搅拌2分钟和在室温下90分钟。添加0.5g活化的离子交换剂(dowex50wx8,200-400目),在室温下搅拌30分钟。然后过滤出离子交换剂,并用甲醇后洗涤。蒸发浓缩滤液,将残余物在真空中干燥。这产生114mg(理论值的95%)的作为阻转异构体混合物的标题化合物。
lc-ms(方法a):rt=0.99min;ms(esipos):m/z=561.3[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.79-7.46(m,7h),6.89(d,1h),5.60(dd,1h),5.22-4.97(m,2h),4.84-4.55(m,1h),4.29(d,1h),4.08-3.73(m,2h),1.44-1.33(m,3h)。
实施例10
a)乙酸(1r)-1-[1-(2-氯-6-氟苯基)-3-({3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}甲基)-1h-1,2,4-三唑-5-基]乙基酯(xiv-b-5)
在用冰冷却的同时,将55µl(0.44mmol)的(r)-(-)-2-乙酰氧基丙酰氯(xii-b)逐滴添加到150mg(0.40mmol)的2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙亚氨酸甲酯(xi)和207µl(1.19mmol)dipea在1.5mlthf中的混合物中。在0℃下30分钟后,添加70mg(0.44mmol)的2-氯-6-氟苯基肼(xiii),然后将该混合物在室温下搅拌过夜。然后将反应混合物在微波中在150℃下加热1小时。将几滴水添加到反应混合物中,经色谱(制备型hplc,洗脱剂:乙腈/水梯度,0.1%甲酸)提纯。将含产物的级分冷冻干燥后,产生162mg(理论值的68%)的作为阻转异构体混合物的标题化合物。
lc-ms(方法a):rt=1.10min;ms(esipos):m/z=603.3[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.83-7.49(m,7h),6.96-6.84(m,1h),5.73(d,1h),5.13(d,2h),4.29(br.s.,1h),4.09-3.76(m,2h),1.79(d,3h),1.54(dd,3h)。
b)2-({1-(2-氯-6-氟苯基)-5-[(1r)-1-羟基乙基]-1h-1,2,4-三唑-3-基}甲基)-5-(4-氯苯基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(i-b-5)
将152mg(0.25mmol)来自步骤a)的化合物和250µl(0.25mmol)1m氢氧化钠溶液在1.5ml甲醇中的混合物在0℃下搅拌2分钟和在室温下90分钟。添加0.5g活化的离子交换剂(dowex50wx8,200-400目),在室温下搅拌30分钟。然后过滤出离子交换剂,并用甲醇后洗涤。蒸发浓缩滤液,将残余物在真空中干燥。这产生137mg(理论值的95%)的作为阻转异构体混合物的标题化合物。
lc-ms(方法a):rt=0.99min;ms(esipos):m/z=561.2[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.83-7.43(m,7h),6.90(d,1h),5.60(dd,1h),5.26-4.92(m,2h),4.84-4.54(m,1h),4.29(d,1h),4.11-3.72(m,2h),1.44-1.33(m,3h)。
实施例11
a)乙酸(1s)-1-{3-({3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}甲基)-1-[4-氟-2-(三氟甲基)苯基]-1h-1,2,4-三唑-5-基}乙基酯(xiv-a-6)
在用冰冷却的同时,将55µl(0.44mmol)的(s)-(-)-2-乙酰氧基丙酰氯(xii-a)逐滴添加到150mg(0.40mmol)的2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙亚氨酸甲酯(xi)和207µl(1.19mmol)dipea在1.5mlthf中的混合物中。在0℃下30分钟后,添加100mg(0.44mmol)的4-氟-2-(三氟甲基)苯基肼(xiii)盐酸盐,在室温下搅拌3小时。然后将反应混合物在微波中在150℃下加热3小时。将几滴水添加到反应混合物中,经色谱(制备型hplc,洗脱剂:乙腈/水梯度,0.1%甲酸)提纯。将含产物的级分冷冻干燥后,产生129mg(理论值的51%)的标题化合物。
lc-ms(方法a):rt=1.14min;ms(esipos):m/z=637.3[m+h]+
1hnmr(dmso-d6,400mhz):δ=8.05-7.54(m,7h),6.88(d,1h),5.75(s,1h),5.09(d,2h),4.38-4.18(m,1h),4.08-3.74(m,2h),1.78(br.s.,3h),1.51(d,3h)。
b)5-(4-氯苯基)-2-({1-[4-氟-2-(三氟甲基)苯基]-5-[(1s)-1-羟基乙基]-1h-1,2,4-三唑-3-基}甲基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(i-a-6)
将119mg(0.19mmol)来自步骤a)的化合物和187µl(0.19mmol)1m氢氧化钠溶液在1.1ml甲醇中的混合物在0℃下搅拌2分钟和在室温下90分钟。添加0.5g活化的离子交换剂(dowex50wx8,200-400目),在室温下搅拌30分钟。然后过滤出离子交换剂,并用甲醇后洗涤。蒸发浓缩滤液,将残余物在真空中干燥。这产生110mg(定量)的标题化合物。
lc-ms(方法a):rt=1.03min;ms(esipos):m/z=595.1[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.96-7.87(m,1h),7.83-7.55(m,6h),6.89(d,1h),5.50(d,1h),5.16-4.94(m,2h),4.69-4.50(m,1h),4.28(br.s.,1h),4.07-3.75(m,2h),1.37(d,3h)。
实施例12
a)乙酸(1s)-1-[1-(2-氯-4-氟苯基)-3-({3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}甲基)-1h-1,2,4-三唑-5-基]乙基酯(xiv-a-7)
在用冰冷却的同时,将55µl(0.44mmol)的(s)-(-)-2-乙酰氧基丙酰氯(xii-a)逐滴添加到150mg(0.40mmol)的2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙亚氨酸甲酯(xi)和207µl(1.19mmol)dipea在1.5mlthf中的混合物中。在0℃下30分钟后,添加85mg(0.44mmol)的2-氯-4-氟苯基肼(xiii)盐酸盐,在室温下搅拌2小时。然后将反应混合物在微波中在150℃下加热3小时。将几滴水添加到反应混合物中,经色谱(制备型hplc,洗脱剂:乙腈/水梯度,0.1%甲酸)提纯。将含产物的级分冷冻干燥后,产生157mg(理论值的66%)的标题化合物。
lc-ms(方法a):rt=1.07min;ms(esipos):m/z=603.0[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.90-7.33(m,7h),6.88(d,1h),5.96-5.47(m,1h),5.10(d,2h),4.29(d,1h),4.11-3.74(m,2h),1.82(br.s.,3h),1.53(d,3h)。
b)2-({1-(2-氯-4-氟苯基)-5-[(1s)-1-羟基乙基]-1h-1,2,4-三唑-3-基}甲基)-5-(4-氯苯基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(i-a-7)
将152mg(0.25mmol)来自步骤a)的化合物和252µl(0.25mmol)1m氢氧化钠溶液在1.5ml甲醇中的混合物在0℃下搅拌2分钟和在室温下90分钟。添加0.5g活化的离子交换剂(dowex50wx8,200-400目),在室温下搅拌30分钟。然后过滤出离子交换剂,并用甲醇后洗涤。蒸发浓缩滤液,将残余物在真空中干燥。这产生140mg(定量)的标题化合物。
lc-ms(方法a):rt=1.00min;ms(esipos):m/z=561.2[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.84-7.54(m,6h),7.42(td,1h),6.89(d,1h),5.51(d,1h),5.06(d,2h),4.72-4.51(m,1h),4.40-4.19(m,1h),4.10-3.74(m,2h),1.38(d,3h)。
实施例13
a)乙酸(1r)-1-[1-(2-氯-4-氟苯基)-3-({3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}甲基)-1h-1,2,4-三唑-5-基]乙基酯(xiv-b-7)
在用冰冷却的同时,将109mg(0.73mmol)的(r)-(-)-2-乙酰氧基丙酰氯(xii-b)逐滴添加到250mg(0.66mmol)的2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙亚氨酸甲酯(xi)和345µl(1.98mmol)dipea在5mlthf中的混合物中。在0℃下30分钟后,添加143mg(0.73mmol)的2-氯-4-氟苯基肼盐酸盐(xiii),然后将该混合物在室温下搅拌过夜。然后将反应混合物在微波中在120℃下加热3小时,经色谱(制备型hplc,洗脱剂:乙腈/水梯度,0.1%甲酸)提纯。将含产物的级分冷冻干燥后,产生177mg(理论值的44%)的标题化合物。
lc-ms(方法a):rt=1.10min;ms(esipos):m/z=603.2[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.96-7.31(m,7h),6.89(d,1h),5.75(s,1h),5.26-4.96(m,2h),4.29(br.s.,1h),4.10-3.76(m,2h),1.82(br.s.,3h),1.53(d,3h)。
b)2-({1-(2-氯-4-氟苯基)-5-[(1r)-1-羟基乙基]-1h-1,2,4-三唑-3-基}甲基)-5-(4-氯苯基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(i-b-7)
将165mg(0.27mmol)来自步骤a)的化合物和275µl(0.27mmol)1m氢氧化钠溶液在3.3ml甲醇中的混合物在室温下搅拌30分钟。将几滴50%的甲酸水溶液添加到反应混合物中,借助制备型hplc(制备型hplc,洗脱剂:乙腈/水梯度,0.1%甲酸)提纯。将含产物的级分冷冻干燥后,产生144mg(理论值的93%)的标题化合物。
lc-ms(方法a):rt=0.96min;ms(esipos):m/z=561.00[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.89-7.54(m,6h),7.42(td,1h),6.90(d,1h),5.51(d,1h),5.06(s,2h),4.72-4.51(m,1h),4.30(d,1h),4.12-3.75(m,2h),1.38(d,3h)。
实施例14
a)乙酸(1s)-1-{3-({3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}甲基)-1-[4-氯-2-(三氟甲基)苯基]-1h-1,2,4-三唑-5-基}乙基酯(xiv-a-8)
在用冰冷却的同时,将55µl(0.44mmol)的(s)-(-)-2-乙酰氧基丙酰氯(xii-a)逐滴添加到150mg(0.40mmol)的2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙亚氨酸甲酯(xi)和207µl(1.19mmol)dipea在1.5mlthf中的混合物中。在0℃下30分钟后,添加107mg(0.44mmol)的4-氯-2-(三氟甲基)苯基肼盐酸盐(xiii),在室温下搅拌3小时。然后将反应混合物在微波中在150℃下加热3小时。将几滴水添加到反应混合物中,经色谱(制备型hplc,洗脱剂:乙腈/水梯度,0.1%甲酸)提纯。将含产物的级分冷冻干燥后,产生125mg(理论值的48%)的标题化合物。
lc-ms(方法a):rt=1.19min;ms(esipos):m/z=653.3[m+h]+
1hnmr(dmso-d6,400mhz):δ=8.23-7.54(m,7h),6.88(d,1h),5.75(s,1h),5.22-4.96(m,2h),4.28(d,1h),4.08-3.72(m,2h),1.78(br.s.,3h),1.51(d,3h)。
b)5-(4-氯苯基)-2-({1-[4-氯-2-(三氟甲基)苯基]-5-[(1s)-1-羟基乙基]-1h-1,2,4-三唑-3-基}甲基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(i-a-8)
将129mg(0.2mmol)来自步骤a)的化合物和200µl(0.2mmol)1m氢氧化钠溶液在1.2ml甲醇中的混合物在0℃下搅拌2分钟和在室温下90分钟。添加0.5g活化的离子交换剂(dowex50wx8,200-400目),在室温下搅拌30分钟。然后过滤出离子交换剂,并用甲醇后洗涤。蒸发浓缩滤液,残余物在真空中干燥。这产生109mg(理论值的90%)标题化合物。
lc-ms(方法a):rt=1.08min;ms(esipos):m/z=611.0[m+h]+
1hnmr(dmso-d6,400mhz):δ=8.15-7.92(m,2h),7.82-7.56(m,5h),6.89(d,1h),5.51(d,1h),5.18-4.96(m,2h),4.64(t,1h),4.29(d,1h),4.10-3.73(m,2h),1.37(d,3h)。
实施例15
a)乙酸(1s)-1-{3-({3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}甲基)-1-[2-氯-4-(三氟甲氧基)苯基]-1h-1,2,4-三唑-5-基}乙基酯(xiv-a-9)
在用冰冷却的同时,将55µl(0.44mmol)的(s)-(-)-2-乙酰氧基丙酰氯(xii-a)逐滴添加到150mg(0.40mmol)的2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙亚氨酸甲酯(xi)和207µl(1.19mmol)dipea在1.5mlthf中的混合物中。在0℃下30分钟后,添加107mg(0.44mmol)的2-氯-4-(三氟甲氧基)苯基肼(xiii),然后将该混合物在室温下搅拌过夜。然后将反应混合物在微波中在100℃下加热3小时。将几滴水添加到反应混合物中,经色谱(制备型hplc,洗脱剂:乙腈/水梯度,0.1%甲酸)提纯。将含产物的级分冷冻干燥后,产生152mg(理论值的57%)的标题化合物。
lc-ms(方法a):rt=1.23min;ms(esipos):m/z=669.1[m+h]+
1hnmr(dmso-d6,400mhz):δ=8.01-7.53(m,7h),6.89(d,1h),5.92-5.60(m,1h),5.11(d,2h),4.39-4.19(m,1h),4.09-3.77(m,2h),1.77(br.s.,3h),1.54(d,3h)。
b)5-(4-氯苯基)-2-({1-[2-氯-4-(三氟甲氧基)苯基]-5-[(1s)-1-羟基乙基]-1h-1,2,4-三唑-3-基}甲基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(i-a-9)
将141mg(0.21mmol)来自步骤a)的化合物和210µl(0.21mmol)1m氢氧化钠溶液在1.3ml甲醇中的混合物在0℃下搅拌2分钟和在室温下90分钟。添加0.5g活化的离子交换剂(dowex50wx8,200-400目),在室温下搅拌30分钟。然后过滤出离子交换剂,并用甲醇后洗涤。蒸发浓缩滤液,残余物在真空中干燥。这产生125mg(理论值的94%)标题化合物。
lc-ms(方法a):rt=1.11min;ms(esipos):m/z=627.3[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.87(d,1h),7.82-7.71(m,4h),7.67-7.51(m,4h),6.89(d,1h),5.54(d,1h),5.07(d,2h),4.75-4.58(m,1h),4.39-4.17(m,1h),4.08-3.74(m,2h),1.40(d,3h)。
实施例16
a)乙酸(1s)-1-{3-({3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}甲基)-1-[2,6-二氯苯基]-1h-1,2,4-三唑-5-基}乙基酯(xiv-a-10)
在用冰冷却的同时,将73µl(0.58mmol)的(s)-(-)-2-乙酰氧基丙酰氯(xii-a)逐滴添加到200mg(0.53mmol)的2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙亚氨酸甲酯(xi)和276µl(1.58mmol)dipea在2mlthf中的混合物中。在0℃下1小时后,添加124mg(0.58mmol)的2,6-二氯苯基肼(xiii)盐酸盐,然后将该混合物在室温下搅拌过夜。然后将反应混合物在微波中在100℃下加热3小时。将几滴水添加到反应混合物中,经色谱(制备型hplc,洗脱剂:乙腈/水梯度,0.1%甲酸)提纯。将含产物的级分冷冻干燥后,产生124mg(理论值的37%)的标题化合物。
lc-ms(方法a):rt=1.08min;ms(esipos):m/z=619.0[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.84-7.52(m,1h),6.89(d,1h),5.78(d,1h),5.13(d,2h),4.41-4.20(m,1h),4.11-3.70(m,2h),1.78(s,3h),1.55(d,3h)。
b)5-(4-氯苯基)-2-({1-(2,6-二氯苯基)-5-[(1s)-1-羟基乙基]-1h-1,2,4-三唑-3-基}甲基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(i-a-10)
将119mg(0.19mmol)来自步骤a)的化合物和190µl(0.19mmol)1m氢氧化钠水溶液在2ml甲醇中的混合物在0℃下搅拌2分钟和在室温下90分钟。添加0.5g活化的离子交换剂(dowex50wx8,200-400目),在室温下搅拌30分钟。然后过滤出离子交换剂,并用甲醇后洗涤。蒸发浓缩滤液,残余物在真空中干燥。这产生110mg(定量)标题化合物。
lc-ms(方法a):rt=1.00min;ms(esipos):m/z=576.9[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.81-7.54(m,7h),6.90(d,1h),5.55(d,1h),5.19-4.96(m,2h),4.63(t,1h),4.30(d,1h),4.08-3.77(m,2h),1.41(d,3h)。
实施例17
a)乙酸(1r)-1-[3-({3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}甲基)-1-(2,6-二氯苯基)-1h-1,2,4-三唑-5-基]乙基酯(xiv-b-10)
在用冰冷却的同时,将73µl(0.58mmol)的(r)-(-)-2-乙酰氧基丙酰氯(xii-b)逐滴添加到200mg(0.53mmol)的2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙亚氨酸甲酯(xi)和276µl(1.58mmol)dipea在2mlthf中的混合物中。在0℃下30分钟后,添加124mg(0.58mmol)的2,6-二氯苯基肼(xiii)盐酸盐,然后将该混合物在室温下搅拌过夜。然后将反应混合物在微波中在150℃下加热3小时。将几滴水添加到反应混合物中,经色谱(制备型hplc,洗脱剂:乙腈/水梯度,0.1%甲酸)提纯。将含产物的级分冷冻干燥后,产生151mg(理论值的46%)的标题化合物。
lc-ms(方法a):rt=1.12min;ms(esipos):m/z=619.2[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.83-7.56(m,8h),6.90(d,1h),5.78(d,1h),5.13(d,2h),4.42-4.12(m,1h),4.06-3.74(m,2h),1.78(s,3h),1.55(d,3h)。
b)5-(4-氯苯基)-2-({1-(2,6-二氯苯基)-5-[(1r)-1-羟基乙基]-1h-1,2,4-三唑-3-基}甲基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(i-b-10)
将141mg(0.23mmol)来自步骤a)的化合物和230µl(0.23mmol)1m氢氧化钠溶液在2.4ml甲醇中的混合物在0℃下搅拌2分钟和在室温下90分钟。添加0.5g活化的离子交换剂(dowex50wx8,200-400目),在室温下搅拌30分钟。然后过滤出离子交换剂,并用甲醇后洗涤。蒸发浓缩滤液,残余物在真空中干燥。这产生128mg(97%理论值的)标题化合物。
lc-ms(方法a):rt=1.01min;ms(esipos):m/z=577.2[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.78-7.57(m,7h),6.90(d,1h),5.55(d,1h),5.08(d,2h),4.63(t,1h),4.40-4.19(m,1h),4.12-3.76(m,2h),1.41(d,3h)。
实施例18
a)乙酸(1s)-1-{3-({3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}甲基)-1-[2-(三氟甲氧基)苯基]-1h-1,2,4-三唑-5-基}乙基酯(xiv-a-11)
在用冰冷却的同时,将55µl(0.44mmol)的(s)-(-)-2-乙酰氧基丙酰氯(xii-a)逐滴添加到150mg(0.40mmol)的2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙亚氨酸甲酯(xi)和207µl(1.19mmol)dipea在3mlthf中的混合物中。在0℃下30分钟后,添加143mg(0.73mmol)的2-三氟甲氧基苯基肼(xiii)盐酸盐,然后将该混合物在室温下搅拌过夜。然后将反应混合物在微波中在120℃下加热3小时,经色谱(制备型hplc,洗脱剂:乙腈/水梯度,0.1%甲酸)提纯。将含产物的级分冷冻干燥后,产生141mg(理论值的56%)的标题化合物。
lc-ms(方法a):rt=1.14min;ms(esipos):m/z=635.3[m+h]+。
b)5-(4-氯苯基)-2-({5-[(1s)-1-羟基乙基]-1-[2-(三氟甲氧基)苯基]-1h-1,2,4-三唑-3-基}甲基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(i-a-11)
将140mg(0.22mmol)来自步骤a)的化合物和220µl(0.22mmol)1m氢氧化钠溶液在2ml甲醇中的混合物在室温下搅拌60分钟。随后,添加17µl的50%甲酸,经色谱(制备型hplc,洗脱剂:乙腈/水梯度,0.1%甲酸)提纯。将含产物的级分冷冻干燥后,产生123mg(理论值的94%)的标题化合物。
lc-ms(方法a):rt=1.04min;ms(esipos):m/z=593.3[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.77-7.51(m,8h),6.89(d,1h),5.54(d,1h),5.06(d,2h),4.63(t,1h),4.41-4.18(m,1h),4.07-3.77(m,2h),1.40(d,3h)。
实施例19
a)乙酸(1r)-1-{3-({3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}甲基)-1-[2-(三氟甲氧基)苯基]-1h-1,2,4-三唑-5-基}乙基酯(xiv-b-11)
在用冰冷却的同时,将109mg(0.73mmol)的(r)-(-)-2-乙酰氧基丙酰氯(xii-b)逐滴添加到250mg(0.66mmol)的2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙亚氨酸甲酯(xi)和345µl(1.98mmol)dipea在5mlthf中的混合物中。在0℃下30分钟后,添加166mg(0.73mmol)的2-三氟甲氧基苯基肼(xiii)盐酸盐,然后将该混合物在室温下搅拌过夜。然后将反应混合物在微波中在120℃下加热3小时,经色谱(制备型hplc,洗脱剂:乙腈/水梯度,0.1%甲酸)提纯。将含产物的级分冷冻干燥后,产生167mg(理论值的39%)的标题化合物。
lc-ms(方法a):rt=1.13min;ms(esipos):m/z=635.3[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.83-7.48(m,8h),6.89(d,1h),5.75(d,1h),5.10(d,2h),4.41-4.20(m,1h),4.11-3.76(m,2h),1.75(s,3h),1.53(d,3h)。
b)5-(4-氯苯基)-2-({5-[(1r)-1-羟基乙基]-1-[2-(三氟甲氧基)苯基]-1h-1,2,4-三唑-3-基}甲基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(i-b-11)
将158mg(0.25mmol)来自步骤a)的化合物和250µl(0.25mmol)1m氢氧化钠溶液在3ml甲醇中的混合物在0℃下搅拌2分钟和在室温下90分钟。随后,添加19µl的50%甲酸,经色谱(制备型hplc,洗脱剂:乙腈/水梯度,0.1%甲酸)提纯。将含产物的级分冷冻干燥后,产生139mg(理论值的94%)的标题化合物。
lc-ms(方法a):rt=1.00min;ms(esipos):m/z=593.00[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.80-7.51(m,8h),6.90(d,1h),5.54(d,1h),5.06(s,2h),4.63(t,1h),4.39-4.21(m,1h),4.08-3.76(m,1h),1.40(d,1h)。
实施例20
a)乙酸(1s)-1-[1-(2,6-二氟苯基)-3-({3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}甲基)-1h-1,2,4-三唑-5-基]乙基酯(xiv-a-12)
在用冰冷却的同时,将37µl(0.29mmol)的(s)-(-)-2-乙酰氧基丙酰氯(xii-a)逐滴添加到100mg(0.26mmol)的2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙亚氨酸甲酯(xi)和138µl(0.79mmol)dipea在2mlthf中的混合物中。在0℃下30分钟后,添加53mg(0.29mmol)的2,6-二氟苯基肼(xiii),然后将该混合物在室温下搅拌过夜。然后将反应混合物在微波中在120℃下加热3小时,经色谱(制备型hplc,洗脱剂:乙腈/水梯度,0.1%甲酸)提纯。将含产物的级分冷冻干燥后,产生130mg(理论值的83%)的标题化合物。
lc-ms(方法a):rt=1.07min;ms(esipos):m/z=587.1[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.91-7.31(m,7h),6.89(d,1h),5.74(d,1h),5.12(d,2h),4.40-4.18(m,1h),4.09-3.74(m,2h),1.80(s,3h),1.53(d,3h)。
b)5-(4-氯苯基)-2-({1-(2,6-二氟苯基)-5-[(1s)-1-羟基乙基]-1h-1,2,4-三唑-3-基}甲基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(i-a-12)
将120mg(0.20mmol)来自步骤a)的化合物和200µl(0.20mmol)1m氢氧化钠溶液在2ml甲醇中的混合物在室温下搅拌60分钟。随后,添加16µl的50%甲酸,经色谱(制备型hplc,洗脱剂:乙腈/水梯度,0.1%甲酸)提纯。将含产物的级分冷冻干燥后,产生110mg(理论值的99%)的标题化合物。
lc-ms(方法a):rt=0.97min;ms(esipos):m/z=545.2[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.86-7.57(m,5h),7.38(s,2h),6.89(d,1h),5.63(d,1h),5.08(s,2h),4.74(t,1h),4.30(d,1h),4.11-3.71(m,2h),1.39(d,3h)。
实施例21
a)乙酸(1s)-1-{3-({3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}甲基)-1-[4-氯-2-(三氟甲氧基)苯基]-1h-1,2,4-三唑-5-基}乙基酯(xiv-a-13)
在用冰冷却的同时,将37µl(0.29mmol)的(s)-(-)-2-乙酰氧基丙酰氯(xii-a)逐滴添加到100mg(0.26mmol)的2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙亚氨酸甲酯(xi)和138µl(0.79mmol)dipea在2mlthf中的混合物中。在0℃下30分钟后,添加53mg(0.29mmol)的4-氯-2-(三氟甲氧基)苯基肼,然后将该混合物在室温下搅拌过夜。然后将反应混合物在微波中在120℃下加热3小时,经色谱(制备型hplc,洗脱剂:乙腈/水梯度,0.1%甲酸)提纯。将含产物的级分冷冻干燥后,产生76mg(理论值的43%)的标题化合物。
lc-ms(方法a):rt=1.17min;ms(esipos):m/z=669.1[m+h]+
1hnmr(dmso-d6,400mhz):δ=8.01-7.53(m,7h),6.88(d,1h),5.74(d,1h),5.10(d,2h),4.29(d,1h),4.10-3.75(m,2h),1.79(s,3h),1.53(d,3h)。
b)5-(4-氯苯基)-2-({1-[4-氯-2-(三氟甲氧基)苯基]-5-[(1s)-1-羟基乙基]-1h-1,2,4-三唑-3-基}甲基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(i-a-13)
将70mg(0.10mmol)来自步骤a)的化合物和105µl(0.10mmol)1m氢氧化钠溶液在1ml甲醇中的混合物在室温下搅拌30分钟。随后,添加8µl的50%甲酸,经色谱(制备型hplc,洗脱剂:乙腈/水梯度,0.1%甲酸)提纯。将含产物的级分冷冻干燥后,产生59mg(理论值的90%)的标题化合物。
lc-ms(方法a):rt=1.12min;ms(esipos):m/z=627.3[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.87-7.56(m,7h),6.92-6.85(m,1h),5.55(d,1h),5.20-4.96(m,2h),4.68(t,1h),4.29(d,1h),4.10-3.73(m,1h),1.40(d,1h)。
实施例22
a)乙酸(1s)-1-[1-(2-氯苯基)-3-({3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}甲基)-1h-1,2,4-三唑-5-基]乙基酯(xiv-a-14)
在用冰冷却的同时,将5.2ml(41.0mmol)的(s)-(-)-2-乙酰氧基丙酰氯(xii-a)逐滴添加到14.1g(37.3mmol)的2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙亚氨酸甲酯(xi)和32.5ml(41.0mmol)dipea在303mlthf中的混合物中。在0℃下1.5小时后,添加7.35mg(0.44mmol)的2-氯苯基肼(xiii)盐酸盐,在室温下搅拌1.5小时。然后将反应混合物在微波中在100℃下加热10小时。然后向反应混合物添加水和乙酸乙酯,并剧烈搅拌。分离相。水相用乙酸乙酯萃取。有机相用饱和氯化铵水溶液洗涤,经硫酸镁干燥并在真空中浓缩。残余物经色谱(硅胶,洗脱剂:环己烷/乙酸乙酯梯度)提纯。旋转蒸发浓缩含产物的级分后,产生10.4mg(理论值的47%)的标题化合物。
lc-ms(方法a):rt=1.09min;ms(esipos):m/z=585.2[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.84-7.49(m,8h),6.89(d,1h),5.75(br.s,1h),5.22-5.00(m,2h),4.4-3.70(m,3h),1.77(brs,3h),1.58-1.44(m,3h)。
b)2-({1-(2-氯苯基)-5-[(1s)-1-羟基乙基]-1h-1,2,4-三唑-3-基}甲基)-5-(4-氯苯基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(i-a-14)
将10.4g(17.7mmol)来自步骤a)的化合物和2.84g(35.5mmol)50%氢氧化钠水溶液在110ml甲醇/水混合物(10:1)中的混合物在0℃下搅拌2分钟和在室温下1小时。将该混合物倒到水中,用1n盐酸水溶液调节至ph7。水相用mtbe萃取。有机相用饱和氯化钠水溶液洗涤,经硫酸镁干燥并在真空中浓缩。这产生10.6g(定量)的标题化合物。
lc-ms(方法2):rt=1.75min;ms(esipos):m/z=543.1[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.94-7.35(m,8h),6.89(d,1h),5.50(d,1h),5.07(d,2h),4.69-3.70(m,4h),1.38(d,3h)。
实施例23
a)乙酸(1r)-1-[1-(2-氯苯基)-3-({3-(4-氯苯基)-5-氧代-4-[(2r)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}甲基)-1h-1,2,4-三唑-5-基]乙基酯(xiv-b-14)
将1.09g(7.3mmol)的(r)-(-)-2-乙酰氧基丙酰氯(xii-b)逐滴添加到2.5g(6.6mmol)2-{3-(4-氯苯基)-5-氧代-4-[(2s)-3,3,3-三氟-2-羟基丙基]-4,5-二氢-1h-1,2,4-三唑-1-基}乙亚氨酸甲酯(xi)和4.6ml(26.4mmol)dipea在65ml二氧杂环己烷中的混合物中。在室温下30分钟后,添加1.3g(7.3mmol)2-氯苯基肼(xiii)。将反应混合物在室温下搅拌2小时,在回流下加热过夜。浓缩反应混合物,经色谱(硅胶,洗脱剂:环己烷/乙酸乙酯梯度)提纯。旋转蒸发浓缩含产物的级分后,产生2.87g(理论值的74%)的标题化合物。
lc-ms(方法a):rt=1.12min;ms(esipos):m/z=585.2[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.90-7.45(m,8h),6.89(d,1h),5.86-5.53(m,1h),5.11(d,2h),4.37-4.22(m,1h),4.11-3.78(m,2h),1.77(brs,3h),1.53(d,3h)。
b)2-({1-(2-氯苯基)-5-[(1r)-1-羟基乙基]-1h-1,2,4-三唑-3-基}甲基)-5-(4-氯苯基)-4-[(2s)-3,3,3-三氟-2-羟基丙基]-2,4-二氢-3h-1,2,4-三唑-3-酮(i-b-14)
将2.94g(5.0mmol)来自步骤a)的化合物和30mg(0.27mmol)碳酸铯在52ml甲醇中的混合物在室温下搅拌过夜。浓缩反应混合物。残余物用乙酸乙酯溶解,用1n盐酸溶液洗涤,然后用饱和氯化钠水溶洗涤。有机相经硫酸镁干燥并在真空中浓缩。这产生2.63g(理论值的96%)的标题化合物。
lc-ms(方法1):rt=0.94min;ms(esipos):m/z=543.0[m+h]+
1hnmr(dmso-d6,400mhz):δ=7.93-7.42(m,8h),6.90(d,1h),5.50(brd,1h),5.07(s,2h),4.59(brs,1h),4.30(brd,1h),4.13-3.76(m,2h),1.38(d,3h)。
用于测量多形体i晶形的式(i)的化合物的x射线衍射图的测量参数:
扫描轴角度
起始位置[°2θ]2.0066
结束位置[°2θ]37.9906
测量温度[℃]25
阳极材料cu
k-α1[å]1.54060
k-α2[å]1.54443
k-β[å]1.39225
k-α2/k-α10.50000
发电机设定40ma,40kv
入射光束单色器聚焦x射线镜
样品旋转是。
表1:2θ角的最大峰值
附图描述:
图1:多形体i晶形的式(i-a-1)的化合物的x射线衍射图。
- 还没有人留言评论。精彩留言会获得点赞!