头孢洛扎的固体形式和制备方法与流程
硫酸头孢洛扎是头孢洛扎的药学上可接受的盐的实例,其为头孢菌素抗细菌剂,并且具有化学名称(6r,7r)-3-[(5-氨基-4-{[(2-氨基乙基)氨基甲酰基]氨基}-1-甲基-1h-吡唑-2-鎓-2-基)甲基]-7-({(2z)-2-(5-氨基-1,2,4-噻二唑-3-基)-2-[(1-羧基-1-甲基乙氧基)-亚氨基]乙酰基}氨基)-8-氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-甲酸盐,或7β-[(z)-2-(5-氨基-1,2,4-噻二唑-3-基)-2-(1-羧基-1-甲基乙氧基亚氨基)乙酰胺基]-3-{3-氨基-4-[3-(2-氨基乙基)脲基]-2-甲基-1-吡唑鎓基(pyrazolio)}甲基-3-头孢烯-4-甲酸盐。其也被称为cxa-101或fr264205。硫酸头孢洛扎的结构如下显示。
头孢洛扎和合成方法描述于美国专利7,129,232中。包含硫酸头孢洛扎和他唑巴坦钠(zerbaxa®)的组合物用于静脉内施用或输注以治疗细菌感染。
目前头孢洛扎的制造方法涉及由柱色谱、纳米过滤和结晶组成的3-步纯化序列,以得到活性药物成分(api)头孢洛扎,其也被称为头孢洛扎的形式2(wo2015/048217和wo2016/109259)。尽管柱色谱和纳米过滤步骤以高纯度得到头孢洛扎的形式2,但整个过程耗时,体积效率低,产率低,并且需要特殊的制造设备。另外,该方法受困于慢过滤速率,这显著降低了该方法的生产率。
仍然需要用于生产硫酸头孢洛扎的简化和改进的制造方法。如本文所述,硫酸头孢洛扎的新的固体形式(形式3)为简化的纯化方法提供了基础。该固体形式,硫酸头孢洛扎的dmac溶剂化物(本文称为“形式3”)能够实现基于结晶的纯化方法,其替代现有的柱色谱和纳米过滤方法。新方法提供了更高产率,溶剂用量的显著降低,更短的循环时间,并利用标准制造设备,由此得到更多产的、更环保和轻便的制造方法。此外,流线型纯化方法减少了总体制造时间,最终节约成本并降低能量损耗。
发明概述
本发明涉及硫酸头孢洛扎的新型固体形式(硫酸头孢洛扎的dmac溶剂化物(形式3))、包含硫酸头孢洛扎dmac溶剂化物(形式3)的组合物、形式3的合成和使用形式3以制备形式2的改进的结晶方法。新型组合物还包括硫酸头孢洛扎固体形式3和/或头孢洛扎的其他结晶和无定形固体形式。
被称为形式3的硫酸头孢洛扎的dmac溶剂化物可以以湿相(在本文中称为“形式3a”)、干相(在本文中称为“形式3b”)或其混合物存在。如本文所用,术语“形式3”是指形式3a、形式3b和/或其混合物。
硫酸头孢洛扎dmac溶剂化物的新型头孢洛扎固体形式(形式3a)可以通过在12.8、17.5、21.7、24.0和24.6的角度(2θ±0.2)处具有一个或多个特征性衍射的x-射线粉末衍射(xrpd)鉴定。此外,固体形式3a的头孢洛扎还可以在9.4、18.9、24.0、25.7和26.9的角度(2θ±0.2)处具有额外的衍射。
硫酸头孢洛扎dmac溶剂化物的新型头孢洛扎固体形式(形式3b)可以通过在11.0、21.0、22.5和24.0的角度(2θ±0.2)处具有一个或多个特征性衍射的x-射线粉末衍射(xrpd)鉴定。
在本发明的第一个实施方案中,硫酸头孢洛扎dmac溶剂化物(形式3a)通过包含以下步骤的方法获得:
a)将头孢洛扎tfa、水、dmac和第一共溶剂合并以形成浆液;
b)过滤浆液以产生滤液;
c)将滤液和硫酸盐源合并;
d)添加第二共溶剂和dmac溶剂化物种子以产生产物;
e)过滤产物以获得硫酸头孢洛扎dmac溶剂化物的湿固体(形式3a)。
在本发明的一个进一步实施方案中,硫酸头孢洛扎dmac溶剂化物(形式3b)通过干燥步骤e)的硫酸头孢洛扎dmac溶剂化物(形式3a)以产生硫酸头孢洛扎dmac溶剂化物的干燥固体(形式3b)而获得。
在一个进一步实施方案中,将碘和黄原酸盐添加至步骤a)的浆液中。在一个实施方案中,所述黄原酸盐是异丙基黄原酸钾盐(pix)、异丙基黄原酸钠盐(six)、乙基黄原酸钾盐、乙基黄原酸钠盐、叔丁基黄原酸钾盐或叔丁基黄原酸钠盐。在一个进一步实施方案中,所述黄原酸盐是异丙基黄原酸钾盐(pix)。
在本发明的第二个实施方案中,硫酸头孢洛扎的固体形式(形式2)通过包含以下步骤的方法获得:
a)将硫酸头孢洛扎dmac溶剂化物(形式3)、乙腈和水合并以产生溶液;
b)将硫酸装入溶液中;
c)过滤溶液并添加硫酸头孢洛扎晶种以产生浆液;
d)将反溶剂添加至浆液中;
e)将碱添加至浆液中以调节ph;
f)将浆液过滤并用溶剂混合物洗涤,以产生硫酸头孢洛扎的湿晶体(形式1);
g)干燥硫酸头孢洛扎的湿晶体以产生硫酸头孢洛扎的固体形式(形式2)。
在该第二个实施方案中,硫酸头孢洛扎dmac溶剂化物的湿固体、硫酸头孢洛扎dmac溶剂化物的干燥固体或其混合物可用于上述步骤a)中。
在一个进一步实施方案中,在步骤g)中的干燥之前,用丙酮洗涤硫酸头孢洛扎的湿晶体(形式1)。
本发明的第三个实施方案是用于制备硫酸头孢洛扎dmac溶剂化物的湿固体(形式3a)的方法,其包含以下步骤:
a)将头孢洛扎tfa、水、dmac和第一共溶剂合并以形成浆液;
b)过滤浆液以产生滤液;
c)将滤液和硫酸盐源合并;
d)添加第二共溶剂和dmac溶剂化物种子以产生产物;和
e)过滤产物以获得硫酸头孢洛扎dmac溶剂化物的湿固体(形式3a)。
在一个进一步实施方案中,在步骤e)中的过滤之后,将硫酸头孢洛扎dmac溶剂化物的湿固体(形式3a)干燥,以产生硫酸头孢洛扎dmac溶剂化物的干燥固体(形式3b)。
在一个进一步实施方案中,在该方法的步骤a)之后,将碘和黄原酸盐添加至步骤a)的浆液中。在一个实施方案中,所述黄原酸盐是异丙基黄原酸钾盐(pix)、异丙基黄原酸钠盐(six)、乙基黄原酸钾盐、乙基黄原酸钠盐、叔丁基黄原酸钾盐或叔丁基黄原酸钠盐。在一个进一步实施方案中,所述黄原酸盐是异丙基黄原酸钾盐(pix)。
本发明的第四个实施方案是用于制备硫酸头孢洛扎的固体形式(形式2)的方法,其包含以下步骤:
a)将硫酸头孢洛扎dmac溶剂化物(形式3)、乙腈和水合并以产生溶液;
b)将硫酸装入溶液中;
c)过滤溶液并添加硫酸头孢洛扎晶种以产生浆液;
d)将反溶剂添加至浆液中;
e)将碱添加至浆液中以调节ph;
f)将浆液过滤并用溶剂混合物洗涤,以产生湿固体(形式1);
g)干燥湿晶体以产生硫酸头孢洛扎的固体形式(形式2)。
这些制造方法可用于制造适于治疗感染的包含固体形式的头孢洛扎的抗生素组合物。在一些实施方案中,可以使用硫酸头孢洛扎的湿固体(形式3a)、硫酸头孢洛扎的干燥固体(形式3b)或其混合物。例如,用于肠胃外施用的包含头孢洛扎的药物组合物可以使用头孢洛扎dmac溶剂化物(形式3a、形式3b或其混合物)通过包含以下步骤的方法从硫酸头孢洛扎(形式2)获得:(a)将头孢洛扎dmac溶剂化物(形式3a、形式3b或其混合物)转化为硫酸头孢洛扎(形式2),(b)形成在水中包含硫酸头孢洛扎(形式2)的头孢洛扎溶液,和(c)将头孢洛扎溶液冻干以获得冻干的硫酸头孢洛扎组合物。冻干的硫酸头孢洛扎组合物可以与他唑巴坦(或其药学上可接受的盐)合并以获得适于在重构后静脉内施用的药物组合物。
附图简述
图1描绘在本文中称为形式3b的干燥固体形式的硫酸头孢洛扎dmac溶剂化物的热重分析(tga)曲线。
图2描绘在本文中称为形式3b的干燥固体形式的硫酸头孢洛扎dmac溶剂化物的差示扫描量热法(dsc)热谱图。
图3是湿相的硫酸头孢洛扎dmac溶剂化物(形式3a)的x-射线粉末衍射图。
图4是干相的硫酸头孢洛扎dmac溶剂化物(形式3b)的x-射线粉末衍射图。
图5描绘由乙腈/水或2-丙醇/水制备的两种不同的硫酸头孢洛扎形式1浆液的过滤时间。
图6a是显示头孢洛扎tfa合成的已知方法的合成方案的实例。
图6b是用于制备头孢洛扎起始原料(被保护的5-氨基-1-甲基吡唑)的合成方案。
发明详述
本发明涉及硫酸头孢洛扎dmac溶剂化物的新型固体形式(形式3),制备此类的方法,以及利用形式3以制备硫酸头孢洛扎的形式1、然后将其转化为形式2(zerbaxa®(头孢洛扎/他唑巴坦))中使用的头孢洛扎api)的制造方法。除非另有说明,本发明的新型固体形式可以是结晶形式、无定形形式或其混合物。
在本发明的第一个实施方案中,硫酸头孢洛扎dmac溶剂化物(形式3a)通过包含以下步骤的方法获得:
a)将头孢洛扎tfa、水、dmac和第一共溶剂合并以形成浆液;
b)过滤浆液以产生滤液;
c)将滤液和硫酸盐源合并;
d)添加第二共溶剂和dmac溶剂化物种子以产生产物;
e)过滤产物以获得硫酸头孢洛扎dmac溶剂化物的湿固体(形式3a)。
在一个实施方案中,第一共溶剂和第二共溶剂独立地选自酮类、c2-c5醇类、腈类、酰胺类、醚类、其他可混溶的溶剂等。在另一个实施方案中,第一共溶剂和第二共溶剂独立地选自乙腈、异丙醇、叔戊醇、1-丙醇、乙醇、叔丁醇、二氧杂环己烷、thf、其他可混溶的溶剂等。在一个实施方案中,第一共溶剂和第二共溶剂独立地选自乙腈、异丙醇和叔戊醇。在另一个优选实施方案中,第一共溶剂和第二共溶剂是乙腈。
在另一个实施方案中,硫酸盐源选自硫酸、硫酸氢铵、硫酸铵、硫酸氢钠、硫酸氢钾、硫酸氢锂、硫酸氢镁、硫酸氢四丁基铵等。在一个进一步实施方案中,硫酸盐源选自硫酸氢铵、硫酸铵、硫酸氢钠、硫酸氢钾、硫酸氢锂和硫酸氢四丁基铵。在一个进一步实施方案中,硫酸盐源选自硫酸氢铵。
在一个进一步实施方案中,硫酸头孢洛扎晶种选自形式3a、形式3b或其混合物。
在一个进一步实施方案中,将碘和黄原酸盐添加至步骤a)的浆液中。在一个实施方案中,所述黄原酸盐是异丙基黄原酸钾盐(pix)、异丙基黄原酸钠盐(six)、乙基黄原酸钾盐、乙基黄原酸钠盐、叔丁基黄原酸钾盐或叔丁基黄原酸钠盐。在一个进一步实施方案中,所述黄原酸盐是异丙基黄原酸钾盐(pix)。
在某些实施方案中,用黄原酸盐和碘处理头孢洛扎tfa用于提供硫酸头孢洛扎dmac溶剂化物(形式3),其具有小于1ppm的钯水平。通常,头孢洛扎tfa含有约100ppm钯。为了将钯水平一致地降低至药学上可接受的水平,首先将头孢洛扎tfa溶解于水或水/有机混合物中,且然后用约0.1至10mol%黄原酸盐(例如,pix或six)和0.1至5mol%碘处理,产生含钯固体的浆液。将这些含钯固体过滤,且所得批料经历结晶,得到硫酸头孢洛扎dmac溶剂化物,其具有少于1ppm的残余钯。
在本发明的一个进一步实施方案中,硫酸头孢洛扎dmac溶剂化物(形式3b)通过干燥步骤e)的硫酸头孢洛扎dmac溶剂化物(形式3a)以产生硫酸头孢洛扎dmac溶剂化物的干燥固体(形式3b)而获得。
在本发明的第二个实施方案中,硫酸头孢洛扎的固体形式(形式2)通过包含以下步骤的方法获得:
a)将硫酸头孢洛扎dmac溶剂化物(形式3)、乙腈和水合并以产生溶液;
b)将硫酸装入溶液中;
c)过滤溶液并添加硫酸头孢洛扎晶种以产生浆液;
d)将反溶剂添加至浆液中;
e)将碱添加至浆液中以调节ph;
f)将浆液过滤并用溶剂混合物洗涤,以产生硫酸头孢洛扎的湿晶体(形式1);
g)干燥硫酸头孢洛扎的湿晶体以产生硫酸头孢洛扎的固体形式(形式2)。
在该第二个实施方案中,硫酸头孢洛扎dmac溶剂化物的湿固体、硫酸头孢洛扎dmac溶剂化物的干燥固体或其混合物可用于上述步骤a)中。
在一个进一步实施方案中,在步骤g)中的干燥之前,用丙酮洗涤硫酸头孢洛扎晶体的湿晶体(形式1)。
在一个进一步实施方案中,硫酸头孢洛扎晶种选自形式1、形式2或其混合物。
在一个实施方案中,反溶剂选自c1-c5醇类、腈类、醚类、其他可混溶的溶剂等。在另一个实施方案中,反溶剂选自乙腈、异丙醇、叔戊醇、1-丙醇、乙醇、叔丁醇、二氧杂环己烷、甲基叔丁基醚(mtbe)、thf、其他可混溶的溶剂等。在一个进一步实施方案中,反溶剂选自乙腈。
在一个实施方案中,碱是无机或有机碱。在一个进一步实施方案中,碱选自胺碱。在一个进一步实施方案中,碱是三乙胺。
在一个实施方案中,溶剂混合物是水和反溶剂的混合物。在一个进一步实施方案中,溶剂混合物是水和乙腈的混合物。
在一个进一步实施方案中,用氮气吹扫使用真空来干燥形式1的湿晶体。
在本发明中,除非另有定义,否则术语“第一”或“第二”用于表明在该方法期间可以多于一次地添加该方法的要素。第一和第二要素(例如“第一共溶剂”和“第二共溶剂”)可以是不同的或相同的。所述术语用于表示在本发明的所述步骤期间第二次添加该要素。
本发明还涉及包含治疗有效量的硫酸头孢洛扎和药学上可接受的载体的药物组合物,其中硫酸头孢洛扎通过包含以下步骤的方法获得:(a)将硫酸头孢洛扎dmac溶剂化物(形式3)转化为硫酸头孢洛扎(形式2);(b)形成包含硫酸头孢洛扎(形式2)的硫酸头孢洛扎溶液,和(c)将硫酸头孢洛扎溶液冻干以获得冻干的硫酸头孢洛扎组合物。在另一个实施方案中,所述药物组合物进一步包含他唑巴坦或其药学上可接受的盐。
术语“有效量”或“治疗有效量”意指将引发研究人员、兽医、医生或其他临床医生所寻求的组织、系统、动物或人的生物学或医学反应的主题化合物的量。
本发明的药物组合物可以包括通过本文所述的方法获得的硫酸头孢洛扎(形式2),与β-内酰胺酶抑制剂诸如他唑巴坦(cas#:89786-04-9)、avibactam(cas#1192500-31-4)、舒巴坦(cas#68373-14-8)和/或克拉维酸(cas#58001-44-8)组合。β-内酰胺酶抑制剂可以以结晶或无定形形式(诸如冻干的他唑巴坦或结晶他唑巴坦(例如,美国专利号8,476,425和5,763,603)被包括,以获得药物组合物。
可以配制通过本发明的方法获得的包含硫酸头孢洛扎(形式2)的药物组合物以通过肠胃外施用(包括皮下、肌肉内和静脉内)施用来治疗感染。在一个具体实施方案中,将本文所述的药物组合物配制用于通过静脉内注射或输注施用。药物抗生素组合物可以包括冻干的单位剂型(例如,小瓶中的粉末)的硫酸头孢洛扎和稳定量的氯化钠(例如,125至500mg氯化钠/1,000mg头孢洛扎活性剂)。单位剂型可以用药学上可接受的载体溶解,且然后静脉内施用。
用于治疗用途的“药学上可接受的载体”是制药领域中众所周知的,且描述于例如remingtonspharmaceuticalsciences,mackpublishingco.(a.r.gennaro编辑1985)中。例如,可以使用在生理ph下的无菌盐水和磷酸盐缓冲盐水。可以在药物组合物中提供防腐剂、稳定剂、染料和甚至调味剂。例如,可以添加苯甲酸钠、山梨酸和对羟基苯甲酸的酯作为防腐剂。同上,在1449。此外,可以使用抗氧化剂和悬浮剂。
“药学上可接受的盐”是指衍生自此类化合物和有机或无机酸(酸加成盐)或有机或无机碱(碱加成盐)的组合的本发明的化合物的盐。药学上可接受的盐的实例包括但不限于例如以下中描述的那些:"handbookofpharmaceuticalsalts,properties,selection,anduse",p.heinrichstahl和camilleg.wermuth(eds.),由vhca(switzerland)和wiley-vch(frg)出版,2002。本发明的化合物可以以游离碱或盐的形式(两种形式都被认为在本发明的范围内)使用。
在一些实施方案中,反应物的添加顺序并不重要。反应物可以同时一起添加至溶剂(例如,单相溶剂、双相水性共溶剂系统等)中,或可替代地,反应物中的一些可以分开添加,且一些在不同时间点一起添加。
用于制备头孢洛扎的目前制造方法,如下面作为方法a所示,涉及由柱色谱、纳米过滤和结晶组成的3-步纯化序列,以得到头孢洛扎。柱色谱和纳米过滤步骤以高纯度得到头孢洛扎,但整个过程耗时,体积效率低,产率低,并且需要特殊的制造设备。此外,结晶步骤体积效率低,并且受困于缓慢过滤速率,显著降低该方法的生产率。
本发明提供了新型固体形式,其用于简化的制造方法中以产生至少同样纯的硫酸头孢洛扎(形式2),其可用作zerbaxa®中的api。如下面作为方法b所示的新的制造方法消除了现有的柱色谱和纳米过滤步骤。该方法导致更高产率,溶剂用量的显著降低,更短的循环时间,并利用标准制造设备。由此提供更多产的、更环保和轻便的制造方法。此外,流线型纯化方法会减少总体制造时间,最终节约成本并降低能量损耗。
此外,新的制造方法(方法b)涉及硫酸头孢洛扎形式1的改进的结晶方法。与方法a相比,改进的结晶方法导致显著更大的颗粒生长,这进而导致过滤速率增加。这最终减少制造时间,节约成本并降低能量损耗。
硫酸头孢洛扎可以以无定形固体形式或以结晶固体形式或以固体形式的混合物存在。头孢洛扎的结晶固体形式可以以一种或多种独特的固体形式存在,其可以另外包含一个或多个当量的水或溶剂(即分别为水合物或溶剂化物)。
如实施例3中所述,硫酸头孢洛扎dmac溶剂化物(形式3)可以通过形成包含头孢洛扎盐、硫酸盐源、dmac、水和共溶剂的头孢洛扎浆液并在有效形成含有头孢洛扎盐的形式3a的头孢洛扎湿饼的条件下维持该溶液而获得。优选将头孢洛扎溶液维持在有效提供固体形式3a的头孢洛扎的期望纯度和产率的温度下。温度范围为约5℃至约20℃。在一个进一步实施方案中,温度为约12℃至约18℃。最优选地,其为约15℃。除了温度之外,种子量(约0.05至约5.0w/w%)和接种后的老化时间(约0.5至约5.0小时)也是可以调节以获得形式3a固体形式的头孢洛扎的参数。用于制备形式3a硫酸头孢洛扎固体形式的特别优选的方法包括分别维持(1.5-3.5):(0.5-1.5):(1.0-3.0)的水/dmac/乙腈(v/v/v)的比率。更优选的是,该比率为2.5水:1.0dmac:2.0乙腈(v/v/v)。干燥后,硫酸头孢洛扎dmac溶剂化物形式3a转化为形式3b。
优选将含有形式3的头孢洛扎溶液维持在有效提供固体形式1的头孢洛扎的期望纯度和产率的温度下。温度为约5℃至约20℃。在一个进一步实施方案中,温度为约10℃至约14℃。最优选地,其为约12℃。除了温度和强酸的量(例如,相对于头孢洛扎的摩尔量提供0.5至2.5摩尔当量且优选1.0摩尔当量的量的硫酸),种子量(例如,0.5至4.0w/w%)和接种后的老化时间(例如,1-5小时)也是可以调节以获得固体形式的头孢洛扎(形式1)的参数。用于制备硫酸头孢洛扎固体形式(形式1)的特别优选的方法包括维持约40:60至约60:40的乙腈/水的比率(v/v)。更优选地,其为乙腈与水的50:50比率(v/v)。
硫酸头孢洛扎固体形式2可以通过使用硫酸头孢洛扎dmac溶剂化物(形式3a或3b)获得。如实施例4中所述,固体形式2的硫酸头孢洛扎可以通过如下来合成:形成包含硫酸头孢洛扎dmac溶剂化物(形式3a或3b)、乙腈、水和反溶剂的硫酸头孢洛扎浆液,在有效形成含有硫酸头孢洛扎形式1的头孢洛扎湿饼的条件下维持该溶液,且然后在干燥后,将硫酸头孢洛扎形式1转化为形式2。
本发明在以下通用方案和随后的实验部分中的实施例中说明。阐述该部分以帮助理解本发明,但并不意图且也不应该被解释为以任何方式限制在此后的权利要求书中所阐述的本发明。
通过在合成方案中所概述的一般方法制备本发明的化合物。
参考本公开,除非另外特别定义,否则本文说明书中使用的技术和科学术语将具有本领域普通技术人员通常理解的含义。
本文可以使用的一些缩写包括:
aq.水性,
boc(boc)n-叔丁氧基羰基,
c.摄氏度,
calc.计算的,
cxa头孢洛扎,
dmacn,n-二甲基乙酰胺,
dmb1,3-二甲氧基苯,
dmb-三苯甲基3,3-二甲基-1-丁醇三苯基甲基,
edc-hcln-(3-二甲基氨基丙基)-n′-乙基碳二亚胺盐酸盐,
equiv.当量(诸当量),
et3n三乙胺,
h小时(诸小时),
hplc高效液相色谱,
ipa异丙基醇;异丙醇,
ktfa三氟乙酸钾,
mecn乙腈,
ms质谱,
mtbe甲基-叔丁基醚,
pd2dba3三(二亚苄基丙酮)二钯(0),
pix异丙基黄原酸钾盐,
pmb4-甲氧基苄基醚,
ppm百万分率,
r.t.(或rt或rt)室温,
six异丙基黄原酸钠盐,
tert-bu叔丁基,
tatd(z)-2-(5-氨基-1,2,4-噻二唑-3-基)-2-(((1-(叔丁氧基)-2-甲基-1-氧代丙-2-基)氧基)亚氨基)乙酸,
tatd-cle(6r,7r)-7-((z)-2-(5-氨基-1,2,4-噻二唑-3-基)-2-(((1-(叔丁氧基)-2-甲基-1-氧代丙-2-基)氧基)亚氨基)乙酰胺基)-3-(氯甲基)-8-氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-甲酸(4-甲氧基苄基)酯,
tdapp三(4-(二甲基氨基)苯基)亚磷酸盐,
tea三乙胺,
tfa三氟乙酸,
thf四氢呋喃,
tlc薄层色谱,
ubt(2-(3-(1-甲基-5-(三苯甲基氨基)-1h-吡唑-4-基)脲基)乙基)氨基甲酸叔丁酯。
实验细节
x-射线粉末衍射(xrpd)
粉末x-射线衍射数据在以bragg-brentano配置进行配置且配备有使用镍滤光片实现的针对kα单色化的cu辐射源的panalyticalx-pertpropw3040系统上获取。采用固定狭缝光学配置用于数据采集。在2和40°2θ之间获取数据。通过将形式3的湿固体样品或干燥粉末样品轻轻压在浅腔零背景硅保持器上来制备样品。保持器用kapton膜覆盖。将图3和4中所示的图案进行背景校正。结果见于图3和4中。
热分析
(tga):热重分析在perkin-elmertga-7热重分析仪上实施。将干燥的形式3粉末的等分试样(~8mg)以5℃/min从25℃加热至240℃,其中在开口盘中以50ml/min进行氮气吹扫。结果见于图1中。
(dsc):tainstruments2920差示扫描量热计用于监测作为温度升高的函数的热事件。将干燥的形式3粉末的等分试样(~5.5mg)以5℃/min从25℃加热至240℃,其中在铝盖盘中以50ml/min进行氮气吹扫。结果见于图2中。
实施例1
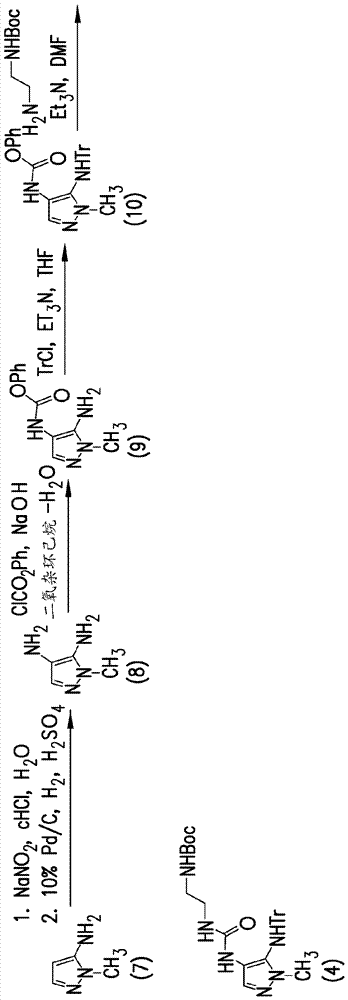
用于制备起始原料以制备头孢洛扎的方法显示于图6a和图6b中,且描述于美国专利号7,129,232和7,192,943,以及toda等人,"synthesisandsarofnovelparenteralanti-pseudomonalcephalosporins:discoveryoffr264205,"bioorganic&medicinalchemistryletters,18,4849-4852(2008)中。
在-10℃下将tatd(70g,在99wt%)和1.0当量的acle-hcl(86.09g,在89.6wt%的游离碱)溶解于525ml乙腈中。添加吡啶(19.91g),随后以3份添加1.2当量的edc∙hcl(48.26g)。将所得反应混合物在-10℃下老化3小时。然后将反应混合物用350ml甲苯和315ml0.5m硫酸溶液淬灭,同时维持内部温度低于-8℃。将双相混合物温热至20-25℃,老化15分钟,且然后使其沉降,以得到两层。将有机层分离并装入315ml0.5m硫酸溶液,同时维持内部温度低于25℃。将双相混合物在25℃下老化15分钟,且然后使其沉降,以得到两层。将有机层分离并装入140ml10wt%氯化钠溶液,并将所得混合物在30℃下老化15分钟。然后使该双相混合物沉降,以得到两层。将所得有机层分离并装入140ml10wt%氯化钠溶液。将反应混合物在30℃下搅拌15分钟,且然后使其沉降,以得到两层。将最终有机层分离并浓缩至455ml的体积。将批料冷却至20℃并装入晶种。将所得浆液在20℃下老化2小时。经8小时向浆液中装入959ml甲苯,且然后在20℃下老化2小时。将晶体收集,用210ml5vol%acn的甲苯溶液洗涤,随后用560ml甲苯洗涤。将滤饼在真空下在25℃下且在氮气吹扫下干燥17小时,以得到tatd-cle。
实施例2
将tatd-cle(70.59g,在85wt%)、ubt(54.77g)、ktfa(20.51g)、tdapp(3.10g)和570mlthf装入反应器中。向反应混合物中装入pd2dba3(0.832g)并在15℃下搅拌15小时。将所得反应混合物用硫代甘油(0.962g)淬灭并浓缩至300ml的体积。通过在蒸馏期间以恒定速率添加540mletoac进行溶剂-转换以除去thf,以维持300ml的恒定体积。在溶剂-转换之后,装入etoac(450ml)、solkafloc(3g)和20wt%氯化钠溶液(180ml)并将反应混合物在15℃下搅拌30分钟。将所得批料过滤,并将有机层分离。添加5wt%nahso4溶液(180ml)并将萃取混合物在15℃下搅拌30分钟。使双相混合物沉降,以得到两层。将有机层分离,用aquaguard(4.5g)装入,并在15℃下搅拌2小时。然后将浆液过滤以除去碳并浓缩至300ml的体积。添加dmb(156ml),并将批料浓缩,以得到300ml的体积。经1小时添加tfa(416ml)并将所得反应混合物在20℃下搅拌3小时。将反应混合物冷却至5℃并装入834mlmtbe,以得到浅褐色浆液。将所得浆液温热至20℃。将固体收集,用mtbe(312ml)洗涤三次,并在真空下用氮气吹扫干燥17小时,以得到94.5g头孢洛扎-tfa(cxa-tfa)。
实施例3
硫酸头孢洛扎dmac溶剂化物(形式3)的制备。
将头孢洛扎-tfa(14.3g,在56wt%)和48ml2.5:1:2水:dmac:乙腈v:v:v(50:19:31w/w)合并,并在15℃下搅拌以得到浆液。以一份添加异丙基黄原酸钾盐(pix)(0.104g,5mol%),并将所得反应浆液在15℃下搅拌30分钟。添加第二份异丙基黄原酸钾盐(0.104g,5mol%),并将反应混合物在15℃下搅拌30分钟。然后添加碘(0.076g,2.5mol%),并将反应浆液在15℃下搅拌1小时。然后将浆液过滤,并将所得废饼用8ml2.5:1:2水:dmac:乙腈洗涤。将滤液和洗涤液合并并冷却至15℃。添加硫酸氢铵(ammoniumbisulfate)(硫酸氢铵(ammoniumhydrogensulfate))(1.449g,1.05当量)并在15℃下搅拌15分钟。添加乙腈(12ml),随后添加硫酸头孢洛扎dmac溶剂化物(形式3)(0.08g)。将所得浆液在15℃下老化3小时。然后经10小时装入乙腈(100ml)并将所得浆液在15℃下老化1小时。将固体过滤,用水:dmac:乙腈(2.5:1:13)洗涤,随后用乙腈洗涤,且然后在真空下用氮气吹扫在25℃下干燥17小时,以得到硫酸头孢洛扎dmac溶剂化物(形式3b)(9.51g,89%产率)。
结晶的硫酸头孢洛扎形式3a(湿相)的x-射线粉末衍射图描绘于图3中,且相应的数据概述于表1中。
表1:硫酸头孢洛扎形式3a的x-射线粉末衍射图
(湿相)图案
结晶的硫酸头孢洛扎形式3b(干相)的x-射线粉末衍射图描绘于图4中,且相应的数据概述于表2中。
表2:硫酸头孢洛扎形式3b的x-射线粉末衍射图
(干相)图案
实施例4
硫酸头孢洛扎(形式2)的制备。
将硫酸头孢洛扎dmac溶剂化物(50g,在70.4wt%)在20℃下溶解于317ml1:1乙腈:水(v:v)中。将批料冷却至15℃,且然后装入50wt%硫酸(10.3g,1.0当量)。将内部温度保持在15-20℃之间(ph=0.1至1.0)。将所得溶液过滤通过0.22μm过滤器,且然后冷却至12℃。向反应混合物中装入硫酸头孢洛扎(形式2或形式1的浆液)(0.35g),并将所得浆液在12℃下老化3小时,以得到种子床。经由注射泵经10小时在12℃下添加乙腈(405ml),且然后老化1小时。装入三乙胺(tea)(5.33g,1.0当量,7.35ml)以调节ph(1.5至3)并将所得浆液老化3小时。将固体过滤,用乙腈:水(4:1)洗涤,随后用丙酮洗涤,以得到硫酸头孢洛扎(形式1),然后将其在真空下用氮气吹扫在25℃下干燥17小时,以得到硫酸头孢洛扎(形式2)(39.3g,95%产率)。
实施例5
用于制备硫酸头孢洛扎形式1的结晶方法:浆液过滤表征
对从ipa/水系统中结晶(如美国专利7,129,232中所述)和从乙腈/水系统中结晶的浆液进行实验室过滤测量。在过滤表征之前,在每种溶剂系统中结晶约30克的形式1水合物。使用rosemund过滤器,以kavon909布(5μm孔隙率)作为过滤介质。压缩氮气用作压力源。使用连接至具有1hz的数据采集的计算机的天平,收集滤液质量作为时间的函数。在实验期间,在乙腈/水中压力从10-40psig变化,并且在实验期间,在ipa/水中压力从10-60psig变化。图5描绘对于从2-丙醇/水和乙腈/水中制备的浆液的压力过滤期间作为时间的函数的滤液重量。如图5中所示,对于等量的形式1晶体(~30g),从乙腈/水中制备(从溶解的形式3中结晶)的形式1材料具有比从2-丙醇/水中制备的形式1材料(~14小时来过滤)显著更短的过滤时间周期(~0.8小时)。与2-丙醇/水相比,从乙腈/水中结晶的材料的更短过滤时间是由于从乙腈/水中制备的较大颗粒以及由于从乙腈/水中的浆液密度增加(即,与ipa/水相比,存在更少的滤液来在从乙腈/水中制备的浆液中除去)。
如本文所述,形式3a和/或3b可以在酸性条件下溶解于乙腈:水中并从乙腈:水中结晶以产生形式1。在干燥后,形式1转化为形式2。在酸性条件下从乙腈/水中形成硫酸头孢洛扎形式1的这种新型结晶方法得到显著更大的颗粒。与现有方法(方法a)相比,这种新型结晶方法也是一种体积生产率更高的方法(~2x改进)。使用乙腈/水结晶的本发明方法产生比2-丙醇(ipa)/水方法显著更大的颗粒。由于更大的颗粒和更高的体积效率,当与从ipa/水中制备的形式1晶体相比时,该方法具有显著更快的过滤速率。从乙腈/水中制备的形式1晶体的改进的体积生产率和更快的过滤速率导致改进的生产能力。
- 还没有人留言评论。精彩留言会获得点赞!