分析并优化基因编辑模块和递送方案的方法与流程
本发明属于产生修饰的细胞系或生物的基因编辑领域。更精确地,本文中报道了一种用于确定基因组整合事件的方法。
发明背景
工程化生物系统和生物的技术是基础科学、药物和生物技术必需的(ran等人,2013)。在最近数年,已经出现几种基因组编辑技术,包括锌指核酸酶(zfn)(miller等人,2007;sander等人,2011;wood等人,2011)、转录激活物样效应核酸酶(talen)(hockemeyer等人,2011;sanjana等人,2012;wood等人,2011;zhang等人,2011)和rna指导的crispr/cas9核酸酶系统(cho等人,2013;cong等人,2013;makarova等人,2011;ran等人,2013)。crispr/cas9核酸酶系统由含有20个与靶dna序列互补的核苷酸的小rna引导(fu等人,2014)。与talen和zfn相反,crispr/cas9系统据称更易设计、有效和充分适用于多种细胞类型和生物的高通量和多重基因编辑(ran等人,2013)。另外,为了产生转基因动物(黑腹果蝇、斑马鱼、小鼠、大鼠和兔),基因编辑技术对于遗传推动的工具(geneticmotivedtools)是必需的,如在细胞系中、原代细胞(体细胞和多潜能干细胞)中和受精卵母细胞中(mcmahon等人,2012;urnov等人,2010)。治疗性基因组编辑的首次应用是在进入门诊的hiv患者的自体cd4t细胞中的ccr5(gori等人,2015;tebas等人,2014)。除了这种应用以外,基因编辑还已经在临床前水平以及在i期临床试验中成功地应用于多种数目的疾病中(cox等人,2015;holt等人,2010;li等人,2011;perez等人,2008;tebas等人,2014;yin等人,2014)。实现基于基因组编辑的疗法的机制是纠正或失活有害突变、引入保护性突变、添加治疗用转基因和破坏病毒dna(cox等人,2015)。因此,基因编辑方案,包括用crispr/cas9技术的方案,可产生全新类别的面向不同疾病的治疗药。
为了治疗性应用(以及研究与开发中的有效使用),表征和比较基因编辑技术及模块是必需的。这包括分析并比较它们的效率和特异性,以及优化向靶细胞的递送。优化递送和评估特异性对基因编辑技术的安全和有效临床转化至关重要(gori等人,2015)。例如,在crispr/cas9系统中迫切想要减少脱靶活性并增加特异性(zhang等人,2015)。脱靶突变可频繁地、以比预期定向突变和插入高得多的比率出现。这可能诱导基因组不稳定性及正常基因的功能的破坏(cho等人,2014;fu等人,2013;mali等人,2013a;pattanayak等人,2013;zhang等人,2015)。目的是优化crispr/cas9系统的多种组分,在不损失中靶(on-target)剪切效率的情况下,促进脱靶活性的降低(zhang等人,2015)。
开发和优化crispr/cas9衍生型应用的一个前提是可靠和稳健地检测及确切地确定杂合基因失活和纯合基因失活以及非特异性整合事件和定向整合事件。现有方法,如确定插入所致表型(例如,药物抗性)或缺少表型(基因失活)或遗传分析/测序方案,往往不区分纯合失活和杂合失活。另外,现有技术很少解析单个细胞的遗传组成,或并不以大量的单个基因编辑细胞为基础来支持稳健的统计分析。
picco,g.等人公开了用于体外和体内选择稳定转导的人细胞的白喉毒素抗性标记物(scientif.rep.5(2015)1-11)。stahl,s.等人公开了白喉酰胺的失去会预激活nf-[κ]b途径和死亡受体途径并且引起mcf7细胞对肿瘤坏死因子超敏感(包括支持信息)(proc.natl.acad.sci.usa112(2015)10732-10737+6p)。
us2016/058889公开,crispr/cas9介导的基因编辑(myo-编辑)有效纠正mdx小鼠(杜兴氏肌营养不良(dmd)模型)中的肌营养不良蛋白基因突变。wo2016/109840公开了使用cas/crispr系统高效率编辑基因组的细胞系、产生这类细胞系的方法、和使用这类细胞系在生物的基因组中产生突变的方法。pradeep,g.k.等人公开,白喉酰胺对延伸因子-2的修饰使得哺乳动物细胞抵抗蓖麻毒蛋白(cell.microbiol.10(2008)1687-1694)。
wo2007/143858公开了dph2基因缺失突变体及其用途。
carette,j.e.等人公开,通过人细胞中的单倍体基因筛选,鉴定被病原体利用的宿主因子(science326(2009)1231-1235)。roy,v.等人公开了防止白喉酰胺形成的显性负向方案可以赋予对假单胞菌外毒素a和白喉毒素的抗性(plosone5(2010)1-7)。
发明简述
本文中报道了一种定量和优化基因编辑方案的稳健和简单方法,该方案基于基因失活与毒素和任选地抗生素选择的组合。该方法不仅可以在大量的独立细胞上确定基因编辑效率,即适用于高通量方法或用途,还允许区分纯合整合事件和杂合整合事件以及区分位点特异性和非特异性的基因破坏和/或整合事件。该方法的简洁性和稳健性使得可以对不同基因编辑模块的效率和特异性进行分析和直接比较,从而鉴定合适的细胞克隆以及整合事件,导致对研究和开发以及治疗应用所需的特性。
一方面,本文报道了一种用于定量crispr/cas介导的基因改变的稳健方法。采用该方法,例如可以确定基因编辑事件的效率、位点特异性和非位点特异性基因破坏之间的差异、以及序列整合事件。因此,如本文所报道的方法可以用于定量和/或优化crisr/cas介导的基因失活和整合事件。
如本文中报道的一个方面是一种用于确定核酸引入哺乳动物细胞的基因组的方法,其中哺乳动物细胞在一个实施方案中包含dph1、dph2、dph4和/或dph5基因的一个或多个转录活性等位基因,在另一个实施方案中包含dph1、dph2、dph4和/或dph5基因的两个转录活性等位基因,所述方法包括步骤
-用一种或多种质粒转染哺乳动物细胞,所述质粒包含待引入的核酸和对所述dph基因进行基因编辑所需的元件,
-在dph基因转录敏感毒素存在下培养转染的细胞,
-如果转染的细胞在毒素存在下存活,则确定核酸引入了哺乳动物细胞的基因组中。
在如本文报道的方法的一个实施方案中,dph基因转录敏感毒素选自假单胞菌外毒素、白喉毒素和cholix毒素。
在如本文中报道的方法的一个实施方案中,该方法包括以下步骤:
-用一种或多种质粒转染哺乳动物细胞,所述质粒包含待引入的核酸、赋予对选择标记的抗性的核酸、和对所述dph基因进行基因编辑所需的元件,
-在选择压力不存在的情况下培养转染的细胞,
-将培养物分成至少两个等分试样,或从培养物取得至少两份样品,和
-在dph基因转录敏感毒素存在下培养第一等分试样或样品,并且在相应的选择标记存在下培养第二等分试样或样品。
如本文所报道的方法可以用来确定基因编辑效率。为了基于统计进行该分析,必须对大量的独立细胞进行独立处理和分析。
在如本文中报道的方法的一个实施方案中,哺乳动物细胞是多数个哺乳动物细胞,并且该方法包括紧在毒素和/或选择标记存在下培养细胞的步骤之前的以下步骤
-将转染的多数个细胞中的各细胞以单细胞的形式分开放置。
在如本文中报道的方法的一个实施方案中,该多数个哺乳动物细胞是1000至10,000,000个细胞。
对获自相同转染的多数个细胞进行的该分析允许确定基因编辑步骤的效率和特异性。
在如本文中报道的方法的一个实施方案中,该方法用于确定基因编辑效率,用于确定基因编辑特异性、或用于确定基因编辑效率和特异性。
由于可以对转染结果实施统计分析,这项分析进一步允许区分纯合基因修饰和杂合基因修饰、以及区分位点特异性和非(位点)特异性基因破坏和整合事件。
在如本文中报道的方法的一个实施方案中,该方法用于确定纯合和杂合基因修饰、或用于确定位点特异性和非(位点)特异性基因破坏和整合。
纯合dph基因失活造成毒素抗性。在这种情况下,dph基因的全部等位基因在细胞中失活。
在如本文中报道的方法的一个实施方案中,如果转染的细胞在选择标记存在下存活,则核酸的引入至少是向哺乳动物细胞的基因组中的单一引入。
毒素敏感性可以区分纯合靶基因修饰和杂合靶基因修饰以及无修饰。因此,如若转染的细胞对毒素敏感但对选择标记不敏感,则转染导致杂合或非特异性基因整合。
在如本文中报道的方法的一个实施方案中,如果转染的细胞在毒素存在下不能存活,但在选择标记存在下存活,则该核酸的引入是向哺乳动物细胞的基因组中引入核酸。
在如本文中报道的方法的一个实施方案中,如果转染的细胞在毒素存在下存活,则核酸的引入是在哺乳动物细胞的基因组中敲除核酸(彻底功能失活)。
对选择标记的抗性表明核酸的整合。如果这是在毒素抗性不存在的情况下,则该整合发生在基因组中与dph基因不同的位置。如果转染的细胞同时地抵抗毒素,则不受理论约束,核酸的整合发生在dph基因中,导致其失活。
在如本文中报道的方法的一个实施方案中,通过在毒素和选择标记存在下培养的组合和任选地高分辨率熔解(hrm)pcr,对dph基因失活事件和核酸整合事件进行定量。
通过比较毒素抗性、选择标记抗性或双重抗性的频率,可以区分整合和整合合并失活。在第一种情况下,这是非(位点)特异性整合(核酸整合,但靶基因未失活),而在第二种情况下,不受理论约束,这是位点特异性整合(核酸整合并且靶基因已经被失活)。纯合基因失活导致毒素抗性;定向核酸整合导致毒素以及选择标记抗性;非定向整合事件仅导致选择标记抗性。
通过比较毒素抗性、选择标记抗性和双重抗性的频率,还可以确定基因编辑方法的特异性和效率。因此,采用如本文中报道的方法,可以评价不同的基因编辑方法并对其排序,或类似地,评价在相同/一种基因编辑方法中使用的不同元件并对其排序。
因此,如本文所报道的方法的稳健读出结果,即毒素抗性集落或选择标记抗性集落的数目,可以用于评价方法变量(例如,crispr/cas基因编辑方法中向导rna的序列长度)对基因修饰效率和修饰类型的影响。
在如本文中报道的方法的一个实施方案中,该方法用于评价/比较不同基因编辑方法的效率并且包括以下步骤
-用一种或多种质粒转染哺乳动物细胞,所述质粒包含待引入的核酸、赋予选择标记抗性的核酸、和用于编辑所述dph基因的第一基因编辑方法所需的元件,
-在选择压力不存的情况下培养转染的细胞,
-将培养物分成至少两个等分试样,或从培养物采取至少两份样品,
-在dph基因表达依赖性毒素存在下培养第一等分试样或样品,并且在相应的选择标记存在下培养第二等分试样或样品,
-对待检验的全部基因编辑方法重复这些步骤,并且
-基于毒素抗性、选择标记抗性或双重抗性的频率,对不同基因编辑方法排序。
白喉毒素对eef2上的白喉酰胺进行adp-核糖基化,由此使eef2失活。这不可逆地停止蛋白质合成并杀死细胞。白喉酰胺是确定的组氨酸修饰,其由白喉酰胺合成基因例如白喉酰胺生物合成基因1、2、4和5(dph1、dph2、dph4和dph5)产生。这些基因的失活将终止毒素靶标的合成并使细胞抵抗假单胞菌外毒素a和白喉毒素(stahl等人,2015)。
可以通过计数毒素抗性集落,以快速和稳健方式检测靶基因的全部等位基因的失活频率。
在如本文中报道的方法的一个实施方案中,通过计数毒素抗性集落,检测细胞中靶基因的全部等位基因(体外)失活的频率。
可以通过对培养的细胞直接进行hrm-pcr测定法,鉴定其中靶基因的仅一个等位基因已经被修饰的细胞。与野生型基因来源的片段相比,对crispr/cas靶位点的修饰将改变相应dph基因来源的pcr片段的解链温度。这可以由hrm谱图中的双相解链曲线反映。
在如本文中报道的方法的一个实施方案中,借助双相解链曲线的存在,通过hrm-pcr检测细胞中靶基因的一个等位基因的(体外)失活。
在如本文中报道的方法的一个实施方案中,对培养的细胞直接进行hrm-pcr。
由于crispr/cas9介导的基因失活事件在细胞中两个(全部)等位基因上罕见相同,因此许多具有彻底基因失活的细胞也将显示双相hrm曲线。这些细胞可以依据它们的毒素抗性表型而区别于单等位基因改变。
在如本文中报道的方法的一个实施方案中,通过计数毒素抗性集落联合hrm-pcr确定的双相解链曲线,检测由crispr/cas9引起的靶基因的全部等位基因失活的频率。
因此,细胞上高通量hrm-pcr测定法和毒素选择(集落计数)测定法的组合能够实现对杂合和纯合dph基因特异性修饰事件的定量。
示例性使用的嘌呤霉素-n-乙酰基-转移酶(pac),由所应用的crispr/cas9质粒的整合盒编码,可以使嘌呤霉素(pm,选择标记)失活并且因此使细胞抵抗pm。由于这是一般原则,因此这适用于通常用于选择阳性转染细胞的任何抗生素。
因此,毒素抗性,例如,假单胞菌外毒素(pe)和白喉毒素(dt)抗性,通过特异性和纯合靶基因失活而产生。
选择标记(例如,pm)抗性是任何整合事件的特征,与整合位置无关。
可以通过将暴露于靶基因特异性向导rna的选择标记(例如,pm)抗性细胞的数目与暴露于乱序非特异性向导rna的细胞的数目比较,解析位点特异性整合与非(位点)特异性整合的频率。
在如本文中报道的方法的一个实施方案中,将编码dph基因特异性crispr/cas9模块的质粒转染入哺乳动物细胞,随后对转染的细胞进行hrm-pcr以及集落计数测定(以检测毒素(dt)抗性细胞和选择标记(pm)抗性细胞),其中该方法用于确定位点特异性整合与非位点特异性整合的频率。
dph基因失活显示出对匹配性向导rna序列的绝对依赖性,而乱序向导rna不产生任何dt抗性集落。这说明,crisprr/cas9/dph向导介导的pac基因整合偏好地发生在dph基因处,但是不具有绝对的特异性。
在如本文中报道的方法的一个实施方案中,dph基因选自dph1基因、dph2基因、dph4基因和dph5基因。
dph1基因、dph2基因、dph4基因和dph5基因中任一者的两个等位基因的失活将赋予绝对毒素抗性。
在如本文中报道的方法的一个实施方案中,该方法包括步骤:确定毒素抗性集落的数目、抗生素抗性集落数目、和毒素与抗生素抗性集落的数目,其中整合事件(抗生素抗性集落数目)和失活事件(毒素抗性集落数目)之间的比率是/反映该方法的特异性。
在如本文中报道的方法的一个实施方案中,该方法用于选择向导rna以实现核酸的crispr/cas9定向整合,其中该方法包括步骤:提供多种不同的向导rna,选择具有整合事件(抗生素抗性集落数目)和失活事件(毒素抗性集落数目)之间比率最高的向导rna。
毒素抗性集落的频率反映靶基因特异性纯合基因失活。同时,评估抗生素抗性集落数目和毒素-抗生素双重抗性集落数目以监测盒整合情况。整合事件(抗生素抗性)和失活事件(毒素抗性)之间的比率可以用作‘特异性指示物’以鉴定发生特异性整合以及同时基因失活事件最少的条件。如果期望定向整合且同时不遭遇大数目的无效(non-productive)靶基因损伤,则这类条件可能有利。
低数值(例如,抗生素抗性集落相对于毒素抗性集落而言少)反映整合相对于同时发生的失活事件而言无效。高数值(抗生素抗性集落更多和/或毒素抗性集落的数目相对减少)反映在crispr/cas9影响的靶基因处的非特异性剪切。
较短的向导rna不仅改善整合效率(抗生素抗性集落的数目总体更高),还改善有效基因编辑(productivegeneediting)和无效基因编辑(non-productivegeneediting)之间的比率(无插入的毒素抗性集落减少)。
在如本文中报道的方法的一个实施方案中,基因编辑方法选自crispr/cas、锌指核酸酶和talen。
可以通过dph基因修饰优化基因编辑模块和参数,并且此后转移到优化其他基因的基因编辑效率或特异性上。
因此,如果目的在于最有效的基因失活,则可以选择20mer,如果想要在无过多破坏性编辑的情况下整合,则可以优选16-18mer。
由于基因失活改变向导rna靶序列,如果目的在于向先前已处理的细胞再施加crispr/cas模块以增加整合效率,则可以使用本文中报道的方法确定最合适的向导rna。由此,仅未改变的基因可以由最初的crispr/cas9组分修饰,而修饰的基因(无整合)不易受使用相同向导rna的所用第一方案影响。
如本文中报道的一个方面是用于鉴定/选择crispr/cas9或zfn或talen(其突变形式或变体)或其他基因编辑模块的方法,所述方法包括以下步骤
-提供/制备一种或多种基因编辑模块的多种变体,
-用如本文中报道的方法确定在整合事件(抗生素抗性集落数目)和失活事件(毒素抗性集落数目)之间的效率和/或最高比率,和
-鉴定/选择具有最高效率和/或最高比率的变体。
如本文中报道的一个方面是用于选择改变(增强或减少)基因编辑模块/方法的效率或特异性的化合物或化合物组合的方法,所述方法包括以下步骤
-提供一种或多种化合物或一种或多种化合物组合,
-任选地,在所述化合物或化合物组合不存在的情况下,用如本文中报道的方法确定在整合事件(抗生素抗性集落数目)和失活事件(毒素抗性集落数目)之间的效率和/或比率,
-在所述化合物或化合物组合存在的情况下,用如本文中报道的方法,对每种所述化合物或化合物组合分别/单独确定在整合事件(抗生素抗性集落数目)和失活事件(毒素抗性集落数目)之间的效率和/或比率,
-鉴定/选择至少一种具有以下效率和/或比率的化合物或化合物组合,所述效率和/或比率与在所述化合物或化合物组合不存在的情况下实施本文所报道的方法而得到的效率和/或比率不同。
如本文中报道的一个方面是一种用于确定化合物浓度及其添加时间点以增强基因编辑方法的效率或特异性并最大限度减少生长抑制或毒性的方法,所述方法包括以下步骤
-提供一种或多种化合物或一种或多种化合物组合,
-任选地,在所述化合物或化合物组合不存在的情况下,用如本文中报道的方法确定在整合事件(抗生素抗性集落数目)和失活事件(毒素抗性集落数目)之间的效率和/或比率,
-在所述化合物或化合物组合存在的情况下,用如本文中报道的方法,在不同的浓度和/或添加时间点,对每种所述化合物或化合物组合分别/单独确定在整合事件(抗生素抗性集落数目)和失活事件(毒素抗性集落数目)之间的效率和/或比率,
-针对所述的至少一种化合物或化合物组合中的每一化合物或化合物组合,分别地鉴定/选择,相比在所述化合物或化合物组合不存在的情况下实施本文所报道的方法得到的效率和/或比率,具有更高效率和/或更高比率的浓度和/或添加时间点。
在先前报道的方法的一个实施方案中,鉴定/选择具有最高效率和/或最高比率的化合物。
发明详述
定义
必须指出,除非上下文另外明确指出,否则如本文中和权利要求中所用,单数形式“一个(a)”、“一种(an)”和“该(an)”包括复数称谓。因此,例如,对“某细胞”的提及包括多数个的此类细胞及本领域技术人员已知的其等同物,等等。同样,术语“a”(或“an”)、“一个或多个”和“至少一个”可以在本文互换使用。
还应指出,术语“包含”、“包括”和“具有”可以互换使用并且包括术语“由……组成”。
本领域技术人员熟知将氨基酸序列(例如,多肽)转化成编码这种氨基酸序列的相应核酸序列的程序和方法。因此,核酸可以以其由独立核苷酸组成的核酸序列来表征并且同样地可以以由其编码的多肽的氨基酸序列来表征。
术语“约”指后随数值的+/-20%范围。在一个实施方案中,术语“约”指后随数值的+/-10%范围。在一个实施方案中,术语“约”指后随数值的+/-5%范围。
假单胞菌外毒素a(pe)、白喉毒素(dt)、cholix毒素(ct)和相关毒素是对真核翻译延伸因子a(eef2)的白喉酰胺残基进行adp-核糖基化的细菌蛋白。使用nad作为辅底物,adp被转移至eef2的白喉酰胺。这使eef2的功能失活。具有adp-核糖基化的eef2的细胞停止其蛋白质合成并因而死亡。pe、dt、ct和相关毒素需要白喉酰胺在eef2上存在以便发生eef2的adp-核糖基化。无白喉酰胺的eef2不能由这些毒素引起adp-核糖基化。
术语“细胞”、“细胞系”和“细胞克隆”互换使用并且指已经向其中引入外源核酸的细胞,包括此类细胞的后代。“细胞”包括“转化体”和“转化的细胞”,其包括原代转化细胞和从其衍生的后代,而无论传代次数是多少。后代可以在核酸内容方面与亲本细胞不完全相同,反而可以含有突变。本文中包括突变后代,所述突变后代具有与在最初转化的细胞中所筛选或选择的功能或生物学活性相同的功能或生物学活性。
“分离的”核酸指已经与其自然环境的组分分离的核酸分子。分离的核酸包括这样的核酸分子,该核酸分子包含在通常含有该核酸分子的细胞中,但是存在于染色体外或与其天然染色体位置不同的染色体位置处。
“质粒”是一种核酸,其提供为了在宿主细胞中表达其所包含的结构基因所需要的全部元件。一般,表达质粒包括包含复制起点的原核质粒扩增单元,例如用于大肠杆菌的原核质粒扩增单元,和选择标记、真核选择标记、以及一个或多个用于表达目的结构基因的表达盒,所述表达盒各自包含启动子、结构基因和包含多聚腺苷化信号的转录终止子。通常将基因表达置于启动子的控制下,并且将这种结构基因称作与该启动子“有效连接”。类似地,如果调节元件调节核心启动子的活性,则调节元件和核心启动子有效连接。术语“质粒”包括例如穿梭质粒和表达质粒以及转染质粒。
“选择标记”是一种核酸,其允许在相应的选择剂存在下特异性选择或反选择携带选择标记的细胞。一般,选择标记将在宿主细胞中赋予药物抗性或弥补代谢缺陷或分解代谢缺陷。选择标记可以是正向、负向或双功能性的。可用的正向选择标记是抗生素耐药基因。这种选择标记允许在相应的选择剂(例如抗生素)存在下正向选择经其转化的宿主细胞。未转化的细胞不能够在培养物中,在选择性条件下,即在选择剂存在下生长或存活。正向选择标记允许选择携带标记的细胞,而负向选择标记允许选择性消除携带标记的细胞。配合真核细胞使用的选择标记包括例如:氨基糖苷磷酸转移酶(aph)基因,例如,潮霉素(hyg)选择标记、新霉素(neo)选择标记和g418选择标记;二氢叶酸还原酶(dhfr)、胸苷激酶(tk)、谷氨酰胺合成酶(gs)、天冬酰胺合成酶、色氨酸合成酶(选择剂为吲哚)、组氨醇脱氢酶(选择剂为组氨醇d)的基因;和赋予嘌呤霉素、博来霉素、腐草霉素、氯霉素、zeocin和麦考酚酸耐药性的核酸。另外的标记基因例如在wo92/08796和wo94/28143中描述了。
发明详述
优化基因编辑的前提是可靠和稳健地检测并区分杂合基因失活和纯合基因失活以及非特异性整合事件和定向整合事件。现有方法,如确定插入所致的表型(例如,抗性)或表型的缺乏(基因失活)或测序方案,往往不区分纯合失活和杂合失活。另外,现有技术很少解析单个独立细胞的遗传组成,或不以大量的独立基因编辑细胞为基础以允许实施稳健统计分析。
由位点特异性核酸酶引起的脱靶效应可能对细胞有毒,并且难以全面预测和监测。而复杂基因组经常含有与预期dna靶相同或高度同源的多个序列,从而导致脱靶活性和细胞毒性(gaj,t.等人,trendsbiotechnol.31(2013)397-405)。
本文中报道了表征基因编辑事件的简单和稳健方法。dph基因失活、毒素处理和(抗生素)选择标记选择的组合,使得可以在非常大数量的独立细胞上确定基因编辑效力。采用该方法,可以区分纯合基因失活和杂合基因失活以及位点特异性整合和非位点特异性整合。如本文所报道的方法的简单性和稳健性可以用于例如优化基因编辑程序和模块以及用于鉴定和比较基因编辑调节物。
已经采用如本文中报道的方法,通过毒素(例如,白喉毒素(dt))和(抗生素)选择标记(例如,嘌呤霉素(pm))选择和高分辨率熔解(hrm)pcr的组合,定量了例如crispr/cas9介导的或锌指核酸酶(zfn)介导的纯合基因失活和杂合基因失活及盒整合事件的效力和特异性。
可以通过hrm-pcr检测总体基因失活频率:纯合dph1或dph2基因失活造成毒素抗性(例如,白喉毒素抗性(dtr)),因此可以依据毒素敏感性区分纯合和杂合dph修饰。选择标记抗性(例如,嘌呤霉素抗性(pmr))由表达盒的整合引起。
靶基因特异性集落计数可以在每个实验的104-105个独立细胞中区分纯合或杂合失活和整合事件,从而在一个实验中评估数百个携带已失活基因的独立克隆。
已经发现,纯合失活(dtr)发生的频率比定向盒整合(dtr&pmr)或非定向整合(使用乱序rna(scrna)的pmr)的频率高约30-50倍。
已经发现,无整合的杂合基因失活发生的频率比整合发生的频率高100倍以上。
相对于整合,对基因失活的偏好与靶序列、基因或染色体位置无关。
已经发现,集落计数可以解析多个变量,包括向导rna(grna)长度、基因编辑用酶的选择、或非同源末端接合(nhej)或同源重组(hr)的调节物。
采用如本文中报道的方法,已经发现20mergrna对失活最有效,而16-18mergrna提供最高的整合效力。
采用如本文中报道的方法,已经发现,就基因失活以及整合而言,未修饰的crispr/cas9的效率两倍于zfn-编辑的,并且比工程化的高保真crispr/cas9效率高5倍。已经发现,对于不同酶,失活事件、非定向整合事件和定向整合事件之间的比率是相似的。
本文中报道的方法还适于解析nhej或hr的调节对编辑效率和特异性的影响。
因此,用于评估以上方面的方法是有益的。
白喉酰胺修饰
stahl,s.等人(proc.natl.acad.sci.usa112(2015)10732-10737)报道了真核翻译延伸因子2(eef2)是高度保守的蛋白质并且对蛋白质生物合成为必需。人eef2的his715处(或在其他物种的相应位置)的白喉酰胺修饰在全部真核生物中及古细菌对应物中是保守的。它由七个基因编码的蛋白质产生。由白喉酰胺生物合成蛋白质1(dph1)、dph2、dph3和dph4(dnajc24)编码的蛋白质,将3-氨基-3-羧丙基(acp)基团接合至eef2。这种中间体由甲基-转移酶dph5转化成白喉素,后者随后由dph6和dph7酰胺化成白喉酰胺。
白喉酰胺修饰的eef2是adp核糖基化性毒素(包括假单胞菌外毒素a(pe)和白喉毒素(dt))的靶。这些细菌蛋白进入细胞,并使用烟酰胺腺嘌呤二核苷酸(nad)作为底物,催化白喉酰胺的adp核糖基化。这种失活的eef2使蛋白质合成停滞并杀死细胞。
在stahl等人中,应用基因特异性锌指核酸酶(zfn),产生dph基因失活的mcf7细胞。
因此,将编码zfn的质粒转染入mcf7细胞。转染后四十八小时,为了能够实现zfn结合、双链断裂和错修复,依据表型选择或依据遗传分析,鉴定突变的细胞。对于表型选择,使细胞暴露于致死剂量的pe(100nm)以杀死其eef2为毒素的底物的全部细胞。在另外48小时后,移除死细胞并且在含有毒素的培养基中增殖培养物。这种程序产生了用针对dph1、dph2、dph4和dph5的zfn转染的细胞的集落。在毒素选择下,在经靶向dph3、dph6和dph7的zfn转染的或经模拟(mock)转染的细胞中,未获得集落。
为了在无毒素选择情况下鉴定mcf7突变体,来自每个转染的各单细胞接受高分辨率熔解(hrm)分析基因。这项技术可以鉴定含有待分析基因的二个不同等位基因的细胞,因为这些细胞产生双相或奇形解链曲线。对具有双相hrm曲线的候选克隆的分析,证实存在不同的dph等位基因序列。该方法给出了这样的克隆,其中对于全部dph基因而言,所述克隆具有失活的一个基因拷贝和另一个有功能的野生型拷贝。
在野生型细胞中,仅可检测到白喉酰胺修饰的eef2,没有eef2未修饰或白喉素修饰或acp修饰的迹象。dph1、dph2、dph4以及dph5基因彻底失活的细胞不含白喉酰胺修饰的eef2。因此,这些基因对白喉酰胺合成是必需的并且其他基因不能补偿它们的失活。dph1、dph2或dph4彻底失活产生仅可检出未修饰的eef2并且不可检出其他修饰形式的细胞。dph5彻底失活产生acp中间体(先前的蛋白质印迹中所用的抗体不识别具有这种中间体的eef2)。在dph1至dph7具有一个失活拷贝和一个功能拷贝的细胞中,主要的eef2种类是白喉酰胺修饰的eef2。
亲本mcf7和全部七种杂合子-失活的mcf7衍生物(dph1-7)的eef2可以由pe引起adp核糖基化。相反,来自具有彻底失活的dph1或dph2或dph4或dph5基因的细胞的eef2不接受adp核糖基化。仅具有白喉酰胺的eef2是adp核糖基化性毒素的底物,而没有修饰(dph1、dph2、dph4)或具有部分修饰(dph5中acp)的eef2则不是。
在正常生长条件下,未观察到dph失活对全部杂合克隆的生长的影响。此外,dph1、dph2或dph4彻底失活并不造成细胞生长或存活力显著减少。dph1、dph2和dph4彻底失活的细胞仅携带未修饰的eef2。因此,仅存在未修饰的eef2本身不会抑制mcf7生长。观察到dph5彻底失活的全部克隆的生长速率降低。
dph1、dph2、dph4或dph5彻底失活的细胞显示tnf敏感性升高。白喉酰胺缺陷型细胞中nf-κb信号转导途径和死亡受体信号转导途径(已知涉及发育并为发育所必需)被预激活。然而,这些细胞是活的,因为途径的诱导未越过足以在无附加刺激的情况下诱导凋亡的阈值。当触发这些预诱导的途径时,预致敏在表型上变得有意义:全部白喉酰胺合成缺陷型细胞(与靶基因敲除无关)均对tnf诱导的凋亡超敏感。这项研究结果表示,白喉酰胺的存在或不存在影响nf-κb途径或死亡受体途径。
dph基因具有在nm_001383.3(dph1)、nc_000001.11(dph2)、nc_000003.12(dph3)、nm_181706.4(dph4)、bc053857.1(dph5)、nm_080650.3(dph6)和nc_000009.12(dph7)中记载的核苷酸序列。通过分离在暴露于致死剂量的假单胞菌外毒素a后的幸存者克隆或通过基于pcr的hrm分析,获得了突变的克隆。
因此,在用于毒素选择的本文方法的一个实施方案中,将细胞在转染后48小时用100nmpe处理并进一步增殖以产生毒素抗性集落,将这些集落分离并从单细胞再克隆,并且为了遗传筛选,产生基因特异性pcr片段并进行hrm(依据双相解链曲线,标示含有突变的克隆)。
在一个实施方案中,通过以下方式评估亲本和突变mcf-7的生长:将10,000个细胞接种在平底96孔板中并在37℃在增湿的5%co2中温育,使细胞在接种后二十四小时暴露于毒素,测定细胞生长,并且进行细胞增殖测定(例如,celltiterglo96aqueousonesolution细胞增殖测定法,promega,根据生产商的说明,其中通过brdu掺入测定法(rochediagnostics,曼海姆frg),在毒素暴露后72小时,解析扩增(dna复制)。
定义(采用自gaj,t.等人,trendsbiotechnol.31(2013)397-405):
crispr/cas(crispr相关)系统:成簇规律间隔的短回文重复序列是含有多个短同向重复序列的基因座,并且向细菌和古细菌提供获得性免疫。crispr系统依赖于crrna和tracrrna用于序列特异地沉默入侵的外来dna。存在三个类型的crispr/cas系统:在ii型系统中,cas9充当rna指导的dna核酸内切酶,该酶在crrna-tracrrna靶识别时切割dna。
crrna:crisprrna与tracrrna碱基配对以形成指导cas9核酸内切酶至互补性dna位点供切割的2-rna结构。
pam:前间区序列相邻基序是在crrna上出现的短核苷酸基序,由cas9特异性识别并且是其切割dna所必需的。
tracrrna:反式激活性嵌合rna是促进crrna加工的非编码rna,是激活rna指导的cas9切割作用所需要的。
dsb:zfn、talen和crispr/cas9作用的产物,双链断裂,是两条dna链均遭切割时出现的dna损伤形式。
hr:同源指导的修复是dsb修复的模板依赖性途径。通过供应含有同源性的供体模板连同位点特异性核酸酶,hdr忠实地在靶向的基因座处插入供体分子。这种方法能够实现单个或多个转基因的插入、以及单核苷酸置换。
nhej:非同源末端接合是将二个断裂的末端连接或接合在一起的dsb修复途径。nhej不利用同源模板进行修复并且因此一般导致在断口部位引入小插入和缺失,经常引起敲除基因功能的移码。
talen:转录激活物样效应核酸酶是foki切割结构域和衍生自tale蛋白的dna结合结构域的融合物。tale含有各自识别单碱基对的多个33–35氨基酸重复结构域。如同zfn,talen诱导定向的dsb,后者激活dna损伤反应途径并且使得定制改变成为可能。
zfn:锌指核酸酶是来自foki限制性核酸内切酶的非特异性dna切割结构域与锌指蛋白的融合物。zfn二聚体诱导刺激dna损伤反应途径的定向dnadsb。经设计的锌指结构域的结合特异性引导zfn至特定基因组位点。
zfnick酶:锌指切刻酶是在二个foki切割结构域之一中含有失活性突变的zfn。zfnic酶仅产生单链dna断口并且诱导hdr而不激活致突变性nhej途径。
基因编辑方法
过去十年期间,已经演进出可以在各种细胞类型和生物中操作实际上任何基因的方案。这项核心技术——常称作‘基因组编辑’——基于对工程化核酸酶的利用,所述核酸酶由融合于非特异性dna切割模块的序列特异性dna结合结构域组成。通过诱导刺激细胞dna修复机制(包括易错非同源末端接合(nhej)和同源性指导的修复(hdr))的定向dna双链断裂(dsb),这些嵌合核酸酶能够实现高效和精确的基因修饰。这种方法的多用性因dna结合结构域的可编程性而被促进。
这些方法的多用性源自能够定制dna结合结构域以识别实际上任何序列的能力。
因此,执行遗传改变的能力主要取决于所设计的蛋白质的dna结合特异性和亲和力(gaj,t.等人,trendsbiotechnol.31(2013)397-405)。
可以构思两种类型的基因特异性操作:非特异性、非定向诱变和定向基因修饰或基因替换。
将未指明的诱变剂靶向一个基因。定向诱变的结果是局部化的序列改变。
通过原始基因拷贝和外源性基因拷贝之间的同源重组,定向基因替换产生局部化的序列变化。在定向基因替换中,目标是在实验室中以一个设计的序列替代一个现有的序列。该设计的序列使得可以引入更微妙和更广泛的改变。产生定向遗传改变经常被称作“基因打靶”(carroll,d.,genetics,188(20111)773–782)。
锌指核酸酶(zfn)和转录激活物样效应核酸酶(talen)以及crispr/cas代表了一类重新界定生物学研究界限的强力工具。这些嵌合核酸酶由连接至非特异性dna切割结构域的可编程、序列特异性dna结合模块组成。通过诱导在特定基因组位置刺激易错非同源末端接合或同源性指导修复的dna双链断口,zfn和talen能够实现类型广泛的基因修饰。基于成簇规律间隔的短回文重复序列(crispr)/cas的rna指导的dna核酸内切酶,依赖于crrna和tracrrna进行dna的序列特异性修饰。存在三个类型的crispr/cas系统:在ii型系统中,cas9充当rna指导的dna核酸内切酶,该酶在crrna-tracrrna靶识别时切割dna。
通过将位点特异性核酸酶与携带基因座特异性同源臂的供体质粒共递送,单个或多个转基因可以有效地整合入内源基因座。除它们在促进hr中的作用之外,位点特异性核酸酶还允许快速产生具有无效表型(nullphenotype)的细胞系和生物;nhej介导的核酸酶所致dsb的修复导致在靶向的位点引入小的插入或缺失,导致通过移码突变敲除基因功能。位点特异性核酸酶还可以诱导大染色体区段的缺失。已经显示这种方法支持大的染色体倒位和易位。最后,通过同步化核酸酶介导的供体dna与染色体靶的切割,已经通过nhej介导的连接,将大的转基因(高达14kb)引入多种内源基因座(gaj,t.等人,trendsbiotechnol.31(2013)397-405)。
nhej介导的核酸酶所致dsb的修复导致高效引入始于断裂位点的可变长度插入/缺失(indel)突变。因此,引入基因编码序列中的dsb的nhej介导的修复经常引起可以导致基因功能敲除的移码突变。
如果供应双链dna“供体模板”,则核酸酶所致dsb的hr可以用来在断口处或该位点附近引入精确的核苷酸替换或多达7.6kb的插入。最新工作还已经显示,寡核苷酸可以配合zfn用来引入精确的改变、小型插入和大型缺失。zfn已经用来引入nhej介导或hr介导的基因改变(joung,j.k.和sander,j.d.,nat.rev.mol.cellbiol.14(2013)49–55)。
一般,将核酸酶编码的基因通过质粒dna、病毒载体或体外转录的mrna递送入细胞。通过电穿孔或基于阳离子脂质的试剂转染质粒dna或mrna,可能有毒性并且限于某些细胞类型。病毒载体也存在局限性,因为它们复杂、难以产生、具有潜在免疫原性,并且涉及额外的监管障碍。整合酶缺陷型慢病毒载体(idlv)是向抵抗转染的细胞类型递送zfn的有吸引力的备选方案。aav是有前景的zfn递送载体,其已经用来增强zfn介导的hr的效率和在体内驱动zfn介导的基因修正。尽管腺病毒载体可以容纳全长talen基因并向人细胞中递送,但携带talen序列的慢病毒质粒载体倾向于在转导后重排(gaj,t.等人,trendsbiotechnol.31(2013)397-405)。
锌指核酸酶(zfn)
将foki核酸内切酶的非特异性切割结构域(n)与锌指蛋白(zfp)组合的锌指核酸酶,提供了向基因组递送位点特异性双链断裂(dsb)的一种通用途径。
锌指(zf)基序的模块结构和zf结构域的模块识别使其可以成为多样的dna识别基序用于设计人工dna结合蛋白。每个zf基序由大约30个氨基酸组成并折叠成ββa结构,所述结构通过保守的cys2his2残基螯合锌离子而稳定化。zf基序通过将α-螺旋插入dna双螺旋的大沟来结合dna。每个指状物主要与dna底物中的三联体结合。相对于每个zf基序的α-螺旋的起点,位置-1、+1、+2、+3、+4、+5和+6处的关键氨基酸残基贡献了大部分的与dna位点的序列特异性相互作用。可以改变这些氨基酸,同时维持剩余氨基酸作为共有主链以产生具有不同三联体序列特异性的zf基序。通过将这些zf基序的几个串联连接以形成zfp,可以实现与更长dna序列的结合。设计的zfp提供强有力的平台技术,因为其他功能如非特异性foki切割结构域(n)、转录激活蛋白结构域(a)、转录阻遏蛋白结构域(r)和甲基化酶(m),可以与zfp融合以分别形成zfn、锌指转录激活蛋白(zfa)、锌指转录阻遏蛋白(zfr)和锌指甲基化酶(zfm)。
foki限制性酶,细菌的iis型限制性核酸内切酶,识别双链体dna中的非回文性五脱氧核糖核苷酸5'-ggatg-3':5'-catcc-3',并在识别位点下游9/13nt进行切割。durai等人提出,可以用识别更长dna序列的其他天然dna结合蛋白或其他设计的dna结合基序替换foki识别结构域,以产生嵌合核酸酶(durai,s.等人,nucl.acidsres.33(2005)5978-5990)。
foki核酸酶作为二聚体发挥作用并且因此必须为每个靶位点设计二个锌指阵列。早期,zfn使用野生型同型二聚体foki结构域,后者可以形成相同单体zfn的不利二聚体。严格异二聚体foki结构域的使用可以减少不利的同型二聚体种类的形成并且因此具有改善的特异性(joung,j.k.和sander,j.d.,nat.rev.mol.cellbiol.14(2013)49–55)。因此,zfn靶位点由foki切割结构域所识别的5–7bp间隔区分隔的二个锌指结合位点组成(gaj,t.等人,trendsbiotechnol.31(2013)397-405)。
用于单杂交遗传选择系统的质粒:在pdb系列质粒上,报道基因,氯霉素乙酰转移酶(cat)或gfp,位于lac衍生性弱启动子(pwk)下游。由zf结合的9bp靶位点位于距转录起点的特定距离。在pa系列质粒上,zf的基因借助编码氨基酸接头的序列与rna聚合酶α-亚单位的片段(rpoa[1–248])融合。rpoa[1–248]–zf融合物与报道分子质粒中的9bp位点结合,可以召集其他rna聚合酶亚基以刺激报道基因的转录(durai,s.等人,nucl.acidsres.33(2005)5978-5990)。
转录激活物样效应核酸酶(talen)
植物病原性黄单胞菌属(xanthomonas)物种转录激活物样(tal)效应子与foki核酸酶的融合物talen,可以成对结合并切割dna。结合特异性由tal效应子中多态性氨基酸重复序列的可定制阵列来决定。
tal效应子进入胞核,与宿主基因启动子中的效应子特异性序列结合并激活转录。它们的靶向特异性由串联的33–35氨基酸重复序列的中心结构域以及后接的20个氨基酸的单个截短重复序列来决定。天然存在的识别位点统一地在前面存在tal效应子活性所必须的一个t(cermak,t.等人,nucl.acidsres.39(2011)e82)。
tale特异性由称作重复-可变双残基(rvd)的二个高变氨基酸决定。类似于锌指,模块化tale重复序列连接在一起以识别邻近dna序列(gaj,t.等人,trendsbiotechnol.31(2013)397-405)。
tal效应子可以与foki核酸酶的催化结构域融合以体内产生用于基因组编辑的定向dna双链断裂(dsb)。由于foki以二聚体形式实施切割,这些tal效应子核酸酶(talen)成对地发挥作用,跨一个间隔区结合相对的靶,在该间隔区上方foki结构域汇聚以造成断裂。在几乎所有细胞中,dsb都可以借助以下二个高度保守的过程之一修复:经常导致小型插入或缺失并且可以用于基因破坏的非同源末端接合(nhej),和可以用于基因插入或替换的同源重组(hr)。
talen或tal效应子构建体的装配包括两个步骤:(i)将重复序列模块装配成1–10个重复的中间阵列和(ii)将该中间阵列连接成主链以产生最终构建体(cermak,t.等人,nucl.acidsres.39(2011)e82)。
talen靶位点由被长度可变(12–20bp)的间隔区序列分隔的二个tale结合位点组成(gaj,t.等人,trendsbiotechnol.31(2013)397-405)。
对于常见的异二聚体靶位点(即,如一般将出现在天然dna序列中的位点),将配对的talen构建体一起转化入靶细胞。
使用合适的限制性核酸内切酶,将靶向目的人基因的成对talen中的一者亚克隆入哺乳动物表达载体。这些酶切割整个talen对并将编码性序列置于启动子的控制下。使用脂质转染胺2000(invitrogen)遵循生产商的操作方案,通过转染,将所产生的质粒引入hek293t细胞。转染后72小时收集细胞(cermak,t.等人,nucl.acidsres.39(2011)e82)。
成簇规律间隔的短回文重复序列/crispr相关蛋白9(crispr/cas9)
天然存在的crispr/casii型系统已经发展成真核细胞的有力基因编辑工具。尤其是,证实可以将crrna和tracrrna并入单一向导rna(sgrna)后,为这种开发铺平了道路。cas9在dna中产生单一双链断口——基因编辑工具的重要特征。该方法利用真核细胞中的dna修复途径提供两种方式以产生遗传变异。第一方式依赖于非同源末端接合(nhej),其连接切口端,但以经常删除少许碱基的方式进行,这可能危害基因产物,或造成致其失活的移码。在第二方式中,使用另一段与靶具有同源性的dna,通过同源指导的修复(hdr),修复受损的等位基因。通过提供可以通过重组插入的dna元件,可以在序列中实现任何类型的插入、缺失或变化。主要的局限性在于需要与靶毗邻的pam。cas9与非预期靶相互作用的脱靶效应是一个需要策略予以预测和防止的问题(rath,d.等人,biochim.117(2015)119-128)。
在ii型crispr/cas系统中,外来dna的短区段(称作“间隔区”)整合在crispr基因组座位内部并且被转录和加工成短crisprrna(crrna)。这些crrna与反式激活性crrna(tracrrna)退火,并指导cas蛋白对致病性dna的序列特异性切割和沉默。最近研究已经显示,cas9蛋白的靶识别需要crrna内部的“种子”序列和crrna结合区上游含有保守二核苷酸的前间区序列邻近基序(pam)序列。已经显示,通过共递送表达cas9核酸内切酶的质粒和必需crrna组件,crispr/cas系统可以方便地运送至人细胞中(gaj,t.等人,trendsbiotechnol.31(2013)397-405)。
确定纯合子和杂合子的小扩增子高分辨率熔解方法
liew,m.等人(clinicalchemistry50(2004)1156–1164)报道了用于确定纯合子和杂合子的一种小扩增子高分辨率熔解方法。因为异双链体改变解链曲线的形状,故容易鉴定杂合子。向未知样品添加15%已知的纯合基因型,可以允许全部三种基因型的解链曲线分离。
异双链体检测染料(lcgreeni)存在下对短pcr产物的解链分析,用来对snp作基因分型。
尽管采用标准技术依据第二、低温解链过渡的存在可以鉴定杂合子,但采用高分辨率方法区分基因型将容易得多。
在lightcycler中采用临床实验室中常用的试剂进行pcr。10μl反应混合物由10-50ng基因组dna、3mmmgcl2、1xlightcyclerfaststartdnamaster杂交探针主混合物、1xlcgreeni、0.5μm正向引物和反向引物,和0.01u/μl大肠杆菌(escherichiacoli)尿嘧啶n-糖基化酶(ung;roche)组成。启动pcr,其中在50℃保持10分钟以借助ung控制污染,且在95℃保持10分钟以激活聚合酶。在85℃和复性温度之间按照编程的20℃/s转变速率,实施快速热循环。
循环后立即在lightcycler上进行解链分析。通过以下方式马上分析二十个样品:首先加热至94℃,冷却至40℃,再次加热至65℃(均按照20℃/s进行),并且随后按0.05℃/s解链并同时连续采集荧光,直至85℃为止。lightcycler软件用来计算衍生解链曲线。
当使用高分辨率熔解时,将扩增的样品在lightcycler中加热到94℃并快速地冷却至40℃。随后转移lightcycler毛细管至hr-1高分辨率仪器并且按0.3℃/s加热。在65℃和85℃之间分析样品,周转时间为1–2分钟。用hr-1软件分析高分辨率熔解数据。在大多数情况下,将荧光对温度的曲线进行归一化。为了与lightcycler数据的直接比较,使用不归一化的衍生曲线。数据输出后,全部曲线均使用microsoftexcel作图。
本文中报道的方法
基因编辑技术是产生基因修饰的工具、细胞(细胞系)或生物所需的。为了适于常规应用,该技术必须具有高特异性和高效率,而与此同时,低水平的非定向基因修饰(即副反应)。
本文中报道了可以用于确定并由此比较/评定基因编辑方法/基因编辑模块的基因编辑特异性、基因编辑效率和/或非定向基因编辑水平的高通量方法。
本文中报道了可以用于定量基因编辑方法/基因编辑模块(例如,crispr/cas)介导的基因改变的稳健高通量方法。该方法适于区分基因编辑方法/基因编辑模块的效率、其位点特异性和非(位点)特异性基因破坏和序列整合事件(例如,定量crisr/cas介导的基因失活和整合事件)。
在如本文中报道的方法的一个实施方案中,基因编辑方法/模块/技术选自crispr/cas、锌指核酸酶和talon核酸酶。
在如本文中报道的方法的一个实施方案中,毒素是酶活性毒素或其片段,其包括但不限于白喉毒素a链、白喉毒素的无结合性活性片段、外毒素a链(来自铜绿假单胞菌(pseudomonasaeruginosa))、蓖麻毒蛋白a链、相思豆毒蛋白a链、蒴莲根毒蛋白a链、α-帚曲菌素、油桐(aleuritesfordii)蛋白、香石竹毒蛋白、垂序商陆(phytolacaamericana)蛋白(papi、papii和pap-5)、苦瓜(momordicacharantia)抑制蛋白、麻疯树毒蛋白、巴豆毒蛋白、肥皂草(sapaonariaofficinalis)抑制蛋白、多花白树毒蛋白、丝林霉素(mitogellin)、局限曲菌素、酚霉素、伊诺霉素和单端孢霉烯类。
示例性实施方案
下文中使用示例性序列和靶举例说明如本文所报道的方法。这不应视为构成对本发明的限制。在此提供仅作为多种备选(即具体实施方案)中之一者和作为本文所报道方法有作用的证据。
在如本文中报道的方法的一个实施方案中,如本文报告的定量和优化基因编辑方案的稳健和简单方法,利用dph基因失活(在优选的实施方案中dph1或dph2基因失活)与白喉毒素和/或嘌呤霉素选择的组合。采用这种方法不仅可以对大量独立细胞(即,高通量地)确定基因编辑效率,还可以区分纯合基因和杂合基因以及区分位点特异性和非(位点)特异性基因破坏和整合事件。
通过白喉毒素(dt)和嘌呤霉素(pm)选择和高分辨率熔解(hrm)pcr的组合,定量了crispr/cas介导的纯合和杂合dph基因失活事件和盒整合事件。可以通过高分辨率熔解hrm-pcr检测dph基因失活,并且纯合dph基因失活造成毒素抗性。因此,毒素敏感性区分纯合和杂合靶基因修饰。pm抗性检测cripr/cas盒的整合。比较dt抗性、pm抗性或双重抗性克隆/集落的频率,已经观察到纯合基因失活(dt抗性)出现的频率比定向盒整合(dt抗性以及pm抗性)或非定向整合事件(pm抗性)高大约30-50倍。无盒整合的杂合靶基因失活(通过hrm-pcr可检出)的出现频率甚至比盒整合的频率更高(>100倍)。相对于盒整合,基因失活的偏向性和更高效率,与crispr/cas9靶序列、染色体位置或靶基因无关。如本文所报道的方法的稳健读出结果(dt抗性或pm抗性集落的数目)可以用于评价crispr/cas9变量(如,向导rna的序列长度)对基因修饰效率和修饰类型的影响。本文呈现的结果也提示,crispr/cas9衍生的疗法可能更适用于基因失活方案,而非需要定向整合事件的应用。
示例性结果
白喉毒素抗性测定法和hrm-pcr定量并且区分纯合和杂合dph1基因失活
白喉毒素(dt)对白喉酰胺进行adp-核糖基化并且因而使真核翻译延伸因子2(eef2)失活。这不可逆地停止蛋白质合成并杀死细胞(weidle等人)。白喉酰胺是白喉酰胺合成基因编码的酶(包括dph1)在eef2上进行的组氨酸修饰。mcf7细胞中彻底失活dph1可以阻止毒素靶标白喉酰胺的合成。这使得细胞抵抗dt(stahl等人)。结果,dph1的全部拷贝的失活将导致‘dt抗性’表型(dtr)。可以通过计数毒素抗性集落,以稳健方式检测这个表型的频率(见图3)。
因为存在一个剩余有功能的dph1等位基因对毒素敏感性而言即足够,所以dtr细胞携带全部dph1等位基因的功能性敲除。可以通过细胞上hrm-pcr测定法,鉴定仅一个等位基因修饰的细胞(图4)。与野生型片段相比,对靶位点的修饰改变dph1-pcr片段的解链温度,从而产生双相hrm曲线。由于crispr-cas9介导的基因失活是独立事件并且因此在两个等位基因上罕见相同,因此具有彻底基因失活的大部分细胞也将显示双相hrm曲线。这些细胞可以依据它们的毒素抗性表型区别于单等位基因改变。
因此,在一个实施方案中,通过(高通量)hrm-pcr和dtr集落计数测定法的组合,定量并区分杂合和纯合失活事件。
选择标记抗性检测并且区分特异性和非特异性整合事件
由当前实施例中所用crispr/cas9供体质粒的整合盒编码的嘌呤霉素-n-乙酰基-转移酶(pac),可以使嘌呤霉素(pm)失活,并使得修饰的细胞抵抗pm(vara等人)。因此,按照对dtr集落所述的相同方式,通过pm抗性测定法检测并定量pac整合(施加pm替代dt作为选择剂,图3b)。与仅因特异性和纯合靶基因失活所导致的dtr相反,pmr的出现与整合位置无关。在一个实施方案中,通过比较双重抗性集落(dtr+pmr)和pmr集落,解析位点特异性整合与非(位点)特异性整合的频率。
比较crispr/cas9所致dph1失活事件和定向整合事件
为了比较靶特异性失活、整合和非靶整合的频率,将编码dph1特异性crispr/cas9模块的质粒(序列参见图1)转染入mcf7细胞。这些细胞随后接受hrm-pcr以及集落计数测定法以检测dt抗性和pm抗性。图5中总结这些测定法的结果,并且下表中显示各个数据集。图5a显示,dph1基因彻底失活(表明全部dph1等位基因功能丧失)以全部已转染细胞的约6%的频率发生(全部细胞的2.5%视为40%的转染效率,参见下文第一表)。dph1失活显示对匹配的grna序列的绝对依赖性:乱序对照rna(scrna)不产生任何dtr集落。图5b中显示hrm鉴定的克隆的频率与dtr集落出现情况的比较。这些分析揭示,单等位基因失活(hrm证实的毒素敏感)的发生频率两倍于双等位基因失活(dtr细胞)。
表:转染细胞的集落计数和表型频率
‘tfeff.’:通过facs分析,监测gfp-报道分子质粒转染mcf7后荧光细胞的频率,确定转染效率。列出了全部细胞中以%计的gfp阳性细胞相对数目。*这些测定法之一展示了不寻常的高gfp阳性和不寻常的facs样式。所有测定的平均转染效率是30%-40%。‘hrm’:高分辨率熔点pcr阳性细胞定义为与野生型细胞相比展示出明确趋异(双相)解链曲线样式的细胞。‘k.o.’表示不携带有功能的dph1或dph2基因拷贝并且因此抵抗dt的细胞的频率。‘int.‘表示携带pac表达盒并因此抵抗pm的细胞的频率。‘a-d’表示独立实验的各样品。
表:grna长度对定向基因失活和盒整合的影响
‘tfeff.’:通过facs分析,监测gfp-报道分子质粒转染mcf7后荧光细胞的频率,确定转染效率。列出了全部细胞中以%计的gfp阳性细胞相对数目。不携带有功能的dph1基因拷贝的细胞抵抗dt。携带pac表达盒的细胞因此抵抗pm。‘a-d’表示独立(四次重复)实验的各样品。
图6显示dtr集落频率和pmr集落频率的比较(二个不同实验,实验1:a-c,实验2:d-f):dph1双等位基因失活,以比介导pm抗性的pac表达盒整合事件,高30-50倍的效力发生(图6c和图6e,不能确定pac整合的位置或合子性(zygosity))。与dph1特异性grna相比,scrna在相同条件下产生少2倍的pmr集落。因此,cas9/dph1-grna介导的整合偏好地发生在dph1基因处,但没有绝对特异性。因此,可以采用如本文中报道的方法验证这种影响。根据在dph1基因中的偏好性整合,采用dph1向导rna获得的许多pmr集落有dt抗性(图6a和图6d)。采用scrna获得的pmr集落均不抵抗dt。因此,cas9介导的基因失活(包括在两个等位基因上的基因失活)高度特异性发生并且频率比定向pac整合高得多(图6c和图6e)。因此,可以采用如本文中报道的方法验证这种影响。定向整合不仅发生频率更低,而且对于由grna限定的位置具有较低的特异性。
对基因编辑的定量以等同方式应用于另一靶基因dph2
还对一个不同基因,dph2基因,使用了如上文概述的相同方案。即,进行cas9诱导的dph2基因修饰。进行这种修饰以显示如本文所报道的方法的一般适用性。
dph2编码一种在不同染色体上具有不同序列的不同酶,然而对白喉酰胺合成而言也是必需的。dph2缺乏使细胞抵抗dt,方式与dph1缺乏相同(stahl等人)。
图6f中显示dph2编辑的结果,随后是dt抗性和pm抗性评估结果:与我们对dph1的观察相符,观察到纯合dph2失活事件比pac表达盒整合具有更高的频率,变化处于相同数量级范围内(dph2失活比pac表达盒整高约90倍)。失活严格依赖于存在关连grna,而盒整合具有位点偏好性,但是对靶基因没有绝对特异性(比较采用scrna的dph2向导的频率)。
因此,为表征dph1修饰所形成的分析原则也可以适用于分析dph2修饰。
通过使用二个不同的基因dph1和dph2,该结果提供了本分析系统可转用于其他基因的证据。
cas9基因操作模块的比较和优化——grna长度
如上文显示,如本文所报道的方法可以解析基因修饰模块组成的总体影响。
图8显示可以怎样评估并比较不同长度的cas9grna的基因失活以及整合的效力和特异性。所用的全部grna均靶向dph1内部的相同序列段,但是长度从14至26个碱基不等(图8d,grna详情在图1中)。记录dtr集落数目以反映靶基因特异性纯合失活。同时,评估pmr抗性数目和dtr-pmr双重抗性数目以监测盒整合。
采用如本文中报道的方法可以证实,grna长度影响基因失活的效力。
已显示20mer赋予最大的dph1失活效力。通过缩短互补段至18或16个碱基或使其延长至26个碱基,采用如本文中报道的方法确定:保留了显著的特异性基因失活功能,尽管效力低于20mer。
缩减grna至小于16个碱基(14mer)降低dph1失活功能至检测水平以下。整合效力(通过计数pmr事件评估)也受grna长度影响。小于16mer的向导rna(14mer)仅产生很少数pmr集落,不超过乱序对照背景水平。观察了16mer,18mer,20mer,22mer,24mer和26mer的定向整合,用16-18mer实现最佳插入效力。
因此采用如本文中报道的方法证实,采用更大的寡核苷酸不实现效力增益,实际上,更大的尺寸显著减少了特异性插入事件。
使用报道的方法,整合事件(pmr)和失活事件(dtr)之间的比率可以计算为‘特异性指示物’以鉴定在基因失活事件最少情况下发生特异性整合的条件。
如果期望定向整合并且不造成过多无效靶基因损伤,则这类条件可能有利。低数值(例如,相对于dtr集落而言,pmr少)反映整合相对于同时发生的失活事件而言是低效的。高数值(更多pmr和/或相对减少的dtr集落数目)反映在cas9靶向的基因处更有效的整合。
图8e显示依赖于grna长度的计算的特异性系数。采用如本文中报道的方法显示,对16-18mer发现最高的插入/失活值,并且对含有20个或更多碱基的向导rna,发现明显的下降。这说明,对于定向基因失活,20mer相当高效(与先前发表的观察结果一致,参见例如cho等人,2013;bauer等人;jinek等人,2012和2013;mali等人,2013)。然而较短的向导rna似乎对整合更高效。较短的向导rna不仅改善整合效力(pmr集落的数目总体更高),还改善有效(productive)基因编辑和无效(non-productive)基因编辑之间的比率(无插入的dtr集落更少)。
不同基因编辑方案的效力和特异性——酶
使用如本文所报道的方法比较了rna指导的cas9的不同变体以及zfn介导的基因编辑的基因失活和整合事件、效力和特异性。
因此,grna长度和组成保持恒定(dph120mer),应用三种不同的编辑酶:
(i)‘spcas9’特指来自化脓性链球菌(streptococcuspyogenes)的cas9核酸酶,该酶可以视为当前的标准应用(参见,例如ran等人,2013,fu等人,2014);
(ii)spcas9-hf1是非特异性dna结合作用降低、脱靶活性降低并且因此被提出具有更高保真性及特异性的spcas9工程化变体(kleinstiver等人);
(iii)zfn介导的编辑模块,其通过设计的锌指介导的蛋白质-核酸相互作用来识别靶序列(miller等人,2011;urnov等人)。
按照与grna分析相同的方式,记录dtr集落以反映定向基因失活,记录pmr集落以监测盒整合(图8f;下表)。
表:用不同基因编辑实体转染的mcf-7细胞的集落计数和表型频率
用编码不同基因组编辑系统(spcas9,spcas9,zfn)的质粒转染mcf-7细胞。spcas9构建体如之前描述。根据kleinstiver等人(kleinstiver等人,2016),spcas9-hf包含n497a/r661a/q695a/q926a置换。同时,grna由scrna并行替换以解析非特异性活性。dph1特异性zfn从sigmaaldrich
比较采用如本文中报道的方法所测定的基因失活和盒整合总效率,发现:
对crispr/spcas9测定到基因失活和盒整合的最高值。
与crispr/spcas9相比,crispr/spcas9-hf削减了定向基因失活事件到小于20%的dtr集落数目。pmr(整合)抗性集落频率和dt-pm双重抗性集落(整合加定向基因失活)频率也降低。
在相同的条件下,zfn模块减少了dtr集落数目到小于用crispr/spcas9观察到的事件的60%。zfn定向失活的效力因此与spcas9相比降低约2倍并且比工程化的spcas9-hf1高约2-3倍。pmr集落频率并未在crispr/spcas9和zfn之间显著差异。与crispr/spcas9相比,zfn的双重抗性集落(盒整合同时基因失活)或多或少地(30%)减少。
计算dtr(靶基因失活)集落/dt+pm双重抗性(靶位点整合)集落的比率,从该公式取得总体效力:crispr/spcas9、crispr/cas9-hf以及zfn产生了按每一个纯合基因失活事件计相同水平(约4x10-3)的定向整合事件。
因此,通过使用如本文所报道的方法,可以显示编辑系统的总体效力是变动的,但是高于靶基因失活背景的盒整合‘特异性’看起来对全部方案并无显著不同。
dna修复调节物对基因编辑效力和特异性的影响
如本文所报道的方法(包括集落测定法以确定在dph基因编辑时的dtr细胞和pmr细胞)也可以用来解析调节dna修复的化合物的影响。
同源重组(hr)的激活物和非同源末端接合(nhej)的抑制物据描述可以调节基因编辑事件和增加整合效力(song等人;ma等人)。
为了展示如本文所报道的方法用于确定dna修复调节物影响编辑效力和特异性的用途,用这类化合物补充根据如本文所报道方法进行的crispr/spcas9/dph1grna(20mer)编辑测定,并对化合物的影响进行定量。
转染基因编辑模块前4小时(‘早添加’)或转染后18小时(‘晚添加’),施加dna连接酶iv抑制物scr7,持续暴露直至转染后72小时。按照相同的方式,添加rad51调节物rs-1(刺激rad51的化合物1)以刺激hr。两种化合物按照不影响mcf7细胞生长或存活力的剂量施加:scr7为1μm和rs-1为8μm,并且当两者组合时1μm+8μm(scr7+rs1)。与未处理的对照(无化合物)相比,确定rs-1的添加增加pmr集落数目约2倍,而不影响采用scrna获得的集落数目(参见下表)。
表:基因编辑期间暴露于scr7和/或rs-1的mcf-7的表型
按限定的数目(接种的细胞数目)接种转染了用于spcas9介导的dph1编辑的质粒的细胞。转染后72小时开始dt/pm选择。值(w、x、y、z)表示在一式四份独立实验中获得的集落。化合物(rs-1、scr7和rs-1+scr7)添加时间点的影响是。处理的样品对比无化合物的样品,pm抗性集落相对于dt抗性集落的显著性差异以*p<0.05、**p<0.01、***p<0.001指示。
为了定量对总体整合效力的影响,采用如本文中报道的方法测定pmr集落(基因整合)相对于dtr集落(基因失活)的百分数(还参见图10)。
基于如本文所报道的方法的结果,已经发现,在早期时间点添加rs-1导致显著更高的整合效率,然而当晚添加时(启动编辑后18小时),不影响整合效率。因此,为rs1介导的hr刺激选择适宜(早期)时间点对有效(productive)编辑是重要的。这确认hr为定向盒整合的驱动因素。
基于如本文所报道的方法的结果,还已经发现,以相似的程度,早期施加scr7显著地增加相对整合数目(图10与上表)。这确认当施用scr7时增强的有效基因编辑的先前观察(参见ma等人)。
基于如本文所报道的方法的结果,已经进一步发现,与6.5%(仅scr7)或6.9%(仅rs-1)相比,两种化合物组合时,pmr相对于dtr的比率是8.1%。然而,该差异/增幅无显著性(p=0.39vs.rs-1单独),这与先前观察相符(pinder等人;gutscher等人)。
应用高通量技术鉴定能够实现基因编辑改善的条件或组分或条件-组分组合
如本文所报道的方法以基于编辑的细胞基因失活为基础,所述失活赋予针对致死性eef2失活毒素如dt或假单胞菌外毒素a或cholix毒素的敏感性。这些测定法的读出结果是抵抗这种adp核糖基化性毒素的细胞的‘集落计数’。
死亡/存活读出结果非常稳健并且基于细胞的集落计数方案是高通量相容的。因此,该分析原则可以适应于高度平行的测定设置并适应于在自动化(机器人)平台上运行。用于基于细胞的测定法中鉴定表型的自动化高通量平台可以以各种样式获得,并且是本领域熟知的。
所述基于dph编辑的分析原则的高通量/自动化应用,可以测量大数量的不同条件或参数或添加物/调节物对基因编辑、尤其对效率和特异性的影响。可以在合成化合物或天然化合物的集合或文库(示例性的而非限制性的)中解析条件或参数或添加物/调节物。这可以鉴定增强基因编辑效率或特异性的化合物和条件。这类化合物和条件尤其可以在结构或功能方面衍生自或类似于直接或间接影响dna重组和修复、核酸结合、蛋白质与核酸相互作用、或与参与核酸重排、代谢、合成或修复的蛋白质相互作用的实体和化合物类别。
展望
基因组编辑已经作为对科学应用最重要的技术出现。然而,其完整潜力仍受目前所使用编辑方案的效力问题和特异性问题限制。本文所报道的方法能够实现简单和稳健地定量和比较编辑所致基因失活和插入的效力和特异性。它允许基于大量独立细胞,确切地确定杂合和纯合基因失活和非特异性与定向整合事件。通过这种方法产生的知识和参数不仅可以改善基因编辑科学,还可以对开发和优化基因编辑具有特别重要性。
如本文所报道的方法的核心原则包括基于编辑的细胞基因失活,所述失活赋予针对致死性eef2失活毒素如dt的敏感性。毒素靶是通过dph基因编码的蛋白质(例如,dph1或dph2)置于eef2上的白喉酰胺。这些基因中任一者的完全功能丧失(两个等位基因)导致‘绝对’毒素抗性,因为这可以防止白喉酰胺合成。因此,毒素抗性特异地指示了两个靶基因的纯合功能性失活。仅使得一个靶等位基因失活的细胞仍是毒素敏感的。可以通过整合盒上赋予抗性的基因分开评估整合事件。通过失活dph基因所致的毒素抗性是‘绝对的’,无任何背景并且独立于毒素的施加量。这是该方法稳健性和简单性的关键因素:可以通过计数毒素抗性集落评估纯合靶基因失活事件的频率,可以通过pm抗性集落评估插入事件,并且可以通过计数抵抗两者的集落评估双重事件。因此,该方法能够实现在大数量的独立细胞上简单和稳健地分别评估各编辑事件。另外(并且与许多现有工具相反,参见,例如fu等人,2014,doench等人;maruyama等人;shalem等人),可以区分纯合和杂合基因失活与整合事件。因此,简单的集落计数反映有效基因编辑(整合)和破坏性基因编辑(无整合的失活)之间的效力和比率。
通过在二个不同dph基因上比较编辑事件(集落频率)的结果证实,该方法可以给出“可通用的”结果。dph1和dph2编码不同的酶,二者独立地对白喉酰胺合成为必需。结果显示,在两个基因上可比的效力、特异性和破坏/整合比率。不同染色体的不同基因上的不同靶序列的可比读出结果说明,通过应用这种方法确定的规则、依赖性和参数可转用以优化其他基因的编辑。
应用dph基因失活作为表征、区分和优化基因编辑效率的稳健方法,不受限于crispr/cas模块。它可以按照相同方式以相同的读出参数适用于其他基因编辑原则,如锌指核酸酶或talens(carroll2011;christian等人,2010;gaj等人,2013;miller等人,2011;urnov等人,2010)。在不同dph靶基因(位于不同染色体)上观察到的相似总体基因失活和整合频率还表明,衍生自这种方法的‘规则’、依赖性和优化参数具有普遍适用性(即,可转用于其他基因)。因此,可以通过dph基因修饰来优化基因编辑模块和参数,并且此后转移以优化其他基因的基因编辑效率或特异性。
采用如本文中报道的方法,可以再确认先前观察结果((cho等人,2013;jinek等人,2012;jinek等人,2013;mali等人,2013b):20mer向导rna对crispr/cas介导的基因打靶是有效的。以20mer观察到最高的总体基因失活和整合频率。与许多其他的方法(doench等人,2014;fu等人,2014;maruyama等人,2015;shalem等人,2014)相反,在如本文报道的方法中,不仅可以评价总体频率,而且可以按照快速、简单和稳健方式对大量独立细胞区分纯合基因失活和杂合基因失活(和非(位点)特异性整合)事件(参见图9)。这使得可以比较有效基因编辑(盒整合)和破坏性基因编辑(无整合的基因失活)的频率和两者之间的比率。
这些分析导致了有意义的观察结果(尽管用20mer实现了最高总体失活频率):以16-18mer向导rna观察到有效和破坏性编辑之间的‘最佳’比率。因此,如果旨在最高效地失活基因,可以选择20mer,其仍可视为现行grna‘标准’(haeussler等人,2016;liao等人,2015;muller等人,2016)。而另一方面,本文呈现的结果显示,对于编辑事件,如果想要在无过多破坏性编辑情况下的整合,可以优选16-18mer。
有意义的是指出,fu等人(fu等人,2014)已经在基因失活实验中评价了短于20mer的grna并且观察到与20mer可比的失活效力,同时脱靶效应减少。他们的分析基于单等位gfp基因失活(每个细胞仅一个靶基因)并且因此不能在二倍体细胞中解析或区分纯合和杂合失活事件。fu等人还不能定量并比较插入事件。数据(基于大量细胞和基于正常染色体编码的人基因的失活)显示,20mer比更短的向导更高效地诱导基因失活。然而,更短的向导是更高效的插入介导物。
具有低的无效基因破坏的最佳整合效率对‘重复方案’可能特别重要,即,目的在于再施加crispr/cas模块至先前处理过的细胞以增加整合效率时。因为基因失活改变向导rna靶序列,仅未改变的基因可以由原始crispr/cas组分修饰,而修饰的基因(无整合)不易受重复方案影响。
确切地确定杂合基因失活和纯合基因失活以及非特异性整合事件和定向整合事件,对开发和优化crispr/cas衍生的疗法特别重要。本文呈现的结果显示,基因失活按照比定向整合高得多(>100倍)的频率发生。甚至纯合失活(两个等位基因均受影响)也比定向整合高30-50倍的频率发生。基因失活显示‘绝对’依赖于向导rna特异性,而整合显示在靶位点处的偏好整合,但没有达到可以被称作‘特异性’整合的程度(靶基因整合相对于非靶基因整合的频率增加2倍)。由此,对要求明确整合事件的应用,必须专门地评价目前可用的crispr/cas衍生的基因编辑技术。
引用的文献:
bauer,d.e.等人,2015,journalofvisualizedexperiments:jove(95).
brooks,s.c.等人,1973,thejournalofbiologicalchemistry248(17):6251-3.
carroll,d.,2011,genetics188(4):773-82.
cho,s.w.等人,2013,naturebiotechnology31(3):230-2.
cho,s.w.等人,2014,genomeresearch24(1):132-41.
christian,m.等人,2010,genetics186(2):757-61.
cong,l.等人,2013,science339(6121):819-23.
cox,d.b.等人,2015,naturemedicine21(2):121-31.
doench,j.g.等人,2014,naturebiotechnology32(12):1262-7.
fu,y.等人,2013,naturebiotechnology31(9):822-6.
fu,y.等人,2014,naturebiotechnology32(3):279-84.
gaj,t.等人,2013,trendsinbiotechnology31(7):397-405.
gori,j.l.等人,2015,humangenetherapy26(7):443-51.
gutschnert等人,cellreports14(2016)1555–1566.
haeussler,m.等人,2016,genomebiology17(1):148.
hockemeyer,d.等人,2011,naturebiotechnology29(8):731-4.
holt,n.等人,2010,naturebiotechnology28(8):839-47.
jinek,m.等人,2012,science337(6096):816-21.
jinek,m.等人,2013,elife2:e00471.
kleinstiver,b.p.等人,nature529(2016)490-495.
li,h.等人,2011,nature475(7355):217-21.
liao,h.k.等人,2015,naturecommunications6.
may等人,rnabiol.13(2016)605-12.
makarova,k.s.等人,2011,naturereviews.microbiology9(6):467-77.
mali,p.等人,2013,naturebiotechnology31(9):833-8.
mali,p.等人,2013,science339(6121):823-6.
maruyama,t.等人,2015,naturebiotechnology33(5):538-42.
mayer,k.等人,2015,internationalj.ofmolecularsciences16(11):27497-507.
mcmahon,m.a.等人,2012,naturemethods9(1):28-31.
miller,j.c.等人,2007,naturebiotechnology25(7):778-785.
miller,j.c.等人,2011,naturebiotechnology29(2):143-8.
muller,m.等人,2016,journalam.societyofgenetherapy24(3):636-44.
pattanayak,v.等人,2013,naturebiotechnology31(9):839-43.
perez,e.e.等人,2008,naturebiotechnology26(7):808-16.
pinder,j.等人,nucl.acidsres.43(2015)9379–9392.
ran,f.a.等人,2013,natureprotocols8(11):2281-308.
sander,j.d.等人,2011,naturemethods8(1):67-u94.
sanjana,n.e.等人,2012,natureprotocols7(1):171-92.
shalem,o.等人,2014,science343(6166):84-7.
song,j.等人,naturecomm.2016-01-01.
stahl,s.等人,2015,proc.natl.acad.sciusa112(34):10732-7.
tebas,p.等人,2014,thenewenglandjournalofmedicine370(10):901-10.
urnov,f.d.等人,2010,genetics11(9):636-46.
vara,j.a.等人,1986,nucleicacidsresearch14(11):4617-24.
weidle,u.h.等人,cancergen.prot.11(2014)25-38.
wood,a.j.等人,2011,science333(6040):307-307.
yin,h.等人,2014,naturebiotechnology32(6):551-3.
zhang,f.等人,2011,naturebiotechnology29(2):149-53.
zhang,g.等人,1996,biochemicalandbiophysicalresearchcommunications227(3):707-11.
zhang,x.h.等人,2015,nucleicacids4:e264.
附图简述
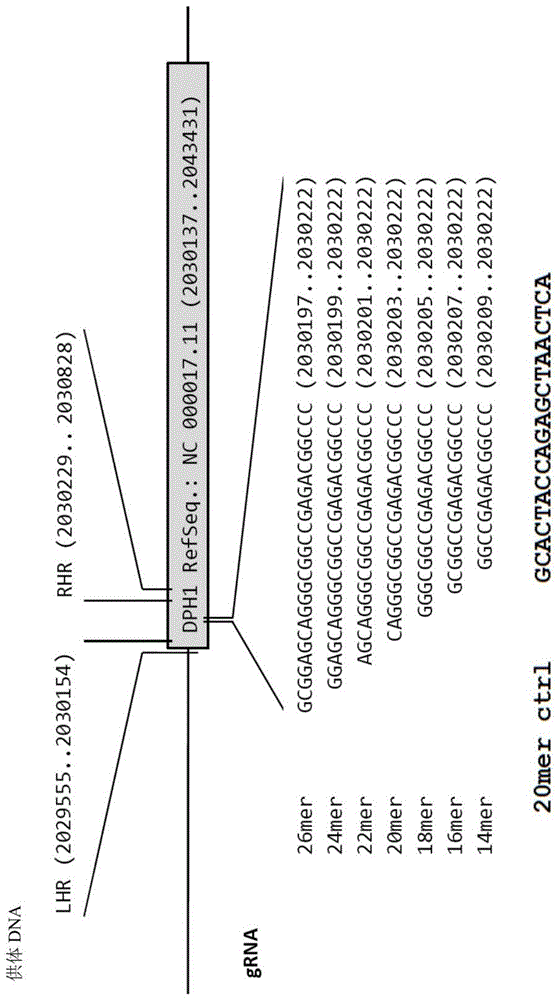
图1grna的组成:靶向dph1(a)和dph2(b)的grna的序列、大小和确切基因组座位;括号中指示确切染色体位置;lhr:供体质粒上整合盒的左侧同源区,rhr:供体质粒上整合盒的右侧同源区。相应的mrna序列是refseq:nm_001383(dph1)和refseq:nm_001039589,nm_001384(dph2)。
图2mcf7细胞和含有杂合(wt/k.o.)或纯合(k.o./k.o.)已失活dph1基因的mcf7暴露于不同浓度的白喉毒素48小时。通过如先前所述的活力测定法评估细胞存活力(mayer等人,2015;stahl等人,2015)。
图3用编码crispr/cas9模块用于失活白喉毒素(dt)敏感性基因dph1以及用于整合赋予嘌呤霉素(pm)抗性的pac表达盒的质粒,转染mcf7细胞。(a&b)转染后48小时,细胞暴露于浓度对含有dph1功能的细胞致死的dt。这种毒素选择产生幸存者集落(a中上方集落),其中全部dph1基因拷贝均失活。保留有功能dph1基因的集落被毒素杀死(a中的集落碎片)。可以通过染色使集落可视化并且将其数目定量为如(b)中所述的集落计数。注意,dt抗性集落仅在用dph1基因特异性向导rna处理细胞时才出现,暴露于对照向导的细胞中无任何非特异性背景。(c)转染后48小时,细胞暴露于浓度对不携带pac表达盒的细胞致死的pm。因此,pm选择产生携带至少一个pac表达盒的幸存者集落。可以通过染色使集落可视化并且将其数目定量为如(c)中所述的集落计数。注意,以用于特异性整合入dph1基因的向导rna处理细胞时,pm抗性集落以更高数目出现,而且在暴露于对照向导的细胞中显示出背景,表明非特异性整合事件。
图4mcf7wt细胞、具有一个dph1野生型等位基因和一个失活等位基因的mcf7wtko细胞、和两个dph1基因均失活的mcf7koko细胞接受hrmpcr。接受hrm的pcr片段覆盖dph1基因中的crispr/cas靶区域。注意,在dph1基因中携带修饰的细胞可以因其解链曲线差异(表示存在改变的等位基因),而区分于野生型细胞。纯合修饰和杂合修饰可以不同于未修饰的野生型细胞。该方法不区分在仅一个等位基因上修饰(杂合、毒素敏感)的细胞和两个等位基因均失活的细胞(纯合、毒素抗性)。毒素敏感性细胞具有表示细胞具有一个野生型等位基因(赋予毒素敏感性)和一个突变等位基因(异常解链点)的双相解链曲线。
图5用dph1特异性crispr/cas模块转染的mcf7细胞接受dt选择,或接受hrm-pcr随后接受dt选择。(a)只有用匹配dph1向导rna,dt选择才产生抗性集落,未处理的细胞中或接受乱序向导rna的细胞中无集落出现。(b)hrm-pcr揭示在其dph1基因的一个或两个等位基因上被修饰的细胞的频率。后续的dt敏感性测定法显示,单等位基因命中(毒素敏感和hrm-阳性)的频率是双等位基因失活(hrm阳性和毒素抗性)的两倍。
图6用dph1特异性crispr/cas模块转染的mcf7细胞接受pm选择和/或接受dt选择。转染对照显示既无dtr集落,也无pmr集落。(a)与乱序向导rna相比,采用dph1向导rna时pm选择产生频率高2倍的抗性集落。在未处理的细胞中无集落出现。(b)pm选择和dt选择的组合可以定量在dph1基因彻底失活的细胞中pac盒整合的频率。dph1向导rna产生具有pm-dt双重抗性的克隆。乱序向导rna仅产生pm抗性克隆,但不产生dt抗性克隆。(c)dt抗性(两个dph1基因均失活)集落频率和pm抗性(pac在dph1基因或在另一个位点整合)频率的比较。(d)显示均值+sem(n=4,***p<0.001)。与乱序向导rna相比,采用dph1grna时pm选择产生频率高2倍的抗性集落。pm选择和dt选择组合揭示了两个dph1等位基因均失活的细胞中pac盒整合的频率。dph1grna产生具有pm-dt双重抗性的克隆。scrna仅产生pmr集落,但不产生dtr集落。(e)显示均值+sem(n=4,***p<0.001)。dtr(两个dph1基因均失活)集落和pmr集落(pac在dph1或在另一个位点整合)的频率比较。(f)显示均值+sem(n=4,***p<0.001)。用dph2特异性grna转染的mcf7细胞接受pm选择和/或接受dt选择。
图7用dph2特异性crispr/cas模块转染的mcf7细胞接受pm选择和/或接受dt选择。按照如对dph1所观察的相同方式,dt抗性集落的频率比pm抗性集落高得多。因此,双等位基因失活比pac盒整合显著更高效。
图8优化基因编辑:grna长度和编辑酶对效力和特异性的影响。(a)采用不同长度的向导rna(grna),用dph1特异性crispr/cas模块转染的mcf7细胞接受pm选择和dt选择。通过读出结果能够确定绝对基因失活频率和盒整合频率、以及确定向导rna无关的非特异性整合事件。(b)通过dt抗性频率和pm抗性频率之间的归一化比率,确定发生所需盒整合并且非整合相关基因失活事件减少的条件。此外注意,用20mer达到破坏性基因编辑的最大效率,而以16-18mer观察到最大整合效率。(c)随18mer向导观察到靶基因处有效整合(抵抗pm以及dt的细胞)的最大效率。(d)显示均值+sem(n=4,λλ/φφ/ψψp<0.01,λλλ/φφφ/ψψψp<0.001)。采用不同长度的grna,用dph1特异性cas9模块转染的mcf-7细胞接受pm选择和dt选择。grna长度影响基因失活和整合频率。所有scrna/grna值均与相应选择组的最大值比较。(e)显示均值+sem(n=4,λλ/φφ/ψψp<0.01,λλλ/φφφ/ψψψp<0.001)。采用不同酶,用dph1特异性cas9模块转染的mcf-7细胞接受pm选择和dt选择。用20mergrna(crispr/cas9)和设计的zfn,dph1编辑中dtr集落、pmr集落或dtr+pmr集落的总数接近。(f)显示均值+sem(n=4,λλ/φφ/ψψp<0.01,λλλ/φφφ/ψψψp<0.001)。采用不同酶,用dph1特异性cas9模块转染的mcf-7细胞接受pm选择和dt选择。总整合事件对总失活事件(pmr/dtr)的比率和位点特异性整合事件/总靶基因失活事件(dtr+pmr)/dtr)的比率。
图9使用靶向人dph1基因的20mer向导rna所观察到的crispr/cas介导的特异性和非特异性基因编辑事件的频率。
图10dna修复调节剂对基因编辑的影响。用编码20mergrna、spcas9和pac的质粒转染mcf-7细胞。在转染之前4小时或在转染后18小时添加hr调节剂rs-1(8μm)和调节nhej的scr7(1μm)。转染后72小时启动dt和/或pm选择。显示了pmr集落(整合)相对于dtr集落(剪切)的百分数。显示均值+sem(n=4)。φ.λp=0.05;φφ,λλp=0.01;φφφλλλp=0.001,相对于未处理的(无添加的药剂)对照。
提供以下实施例、附图和序列以辅助理解本发明,本发明的范围在所附权利要求书中阐述。可以理解,可以对所述方法作出修改而不脱离本发明的精神。
实施例
实施例1
培养mcf7细胞和转染编码基因编辑实体的质粒
mcf7细胞(brooks等人,1973)最初从atcc获得(随后在自有细胞库中增殖)并且在37℃和85%湿度于补充10%fcs、2mml-谷氨酰胺和青霉素/链霉素的rpmi1641培养基中维持。为了转染携带基因编辑模块的质粒,将3,000,000个细胞接种在10cm直径培养皿中并在37℃和增湿的5%co2中培养。接种后24小时,使用jetpei(polyplus),根据生产商的操作方案,但n/p比率为6:1,用20μg总dna转染细胞。24小时后,通过流式细胞术(facscalibur,bdbiosciences)测定经修饰的egfp表达质粒转染的细胞的转染效率(zhang等人,1996)。
从origene获得编码crispr/cas9基因编辑实体的质粒(即敲除和整合系统),所述实体针对dph1(grna靶序列:cagggcggccgagacggccc(seqidno:01),来自参考序列:nm_001383)和dph2(grna靶序列:gatgtttagcagccctgccg(seqidno:02),来自参考序列:nm_001039589,nm_001384),和乱序对照(grna序列:gcactaccagagctaactca(seqidno:03))。该系统包含在u6启动子控制下表达约100ntgrna以及在cmv启动子控制下表达cas9核酸酶的一个质粒、以及具有旁侧分布有靶基因(dph1或dph2)的同源臂的少启动子gfp/pm表达盒的供体质粒。不同大小的其他dph1grna序列变体(origene)是
(参见图1a)。
实施例2
鉴定和定量crispr/cas9介导的dph1和dph2基因纯合敲除
使得dph1或dph2的全部染色体拷贝均失活的mcf7细胞抵抗白喉毒素(stahl等人,2015)。因此,当crispr/cas所致基因失活时毒素抗性细胞/集落的出现和频率提供了失活全部基因拷贝的效率的量度。因此,如实施例1中所述用(i)作为转染对照的gfp表达质粒、(ii)crispr/cas9dph1或dph2敲除/整合系统和(iii)含有乱序grna的敲除/整合实体,转染mcf/细胞,以确定cas9非依赖性整合频率。在确定转染效率后,将细胞接种在6孔板中。为了通过dt定量纯合敲除事件,接种20000个细胞,并且为了通过pm或两者定量整合事件,接种40000个细胞。接种细胞后3天,将rpmi培养基用含有dt或pm或两种毒素的rpmi培养基替换。每2–3天重复培养基的交换,直至从平板移除全部死细胞并且可以通过显微镜检查检测到活跃生长的集落。此后(在毒素暴露开始后第12天和第14天之间),将细胞用pbs洗涤3次并随后用冰冷的亚甲基蓝(50%etoh中的0.2%)染色。随后移除亚甲基蓝溶液并且在流水下小心地洗涤平板。最后,显微镜下用5mm网格箔计数细胞集落。这些分析的结果汇总于下表中。
表:已转染的mcf-7细胞的集落计数和表型频率
‘tfeff.’:通过facs分析,监测gfp-报道分子质粒转染mcf-7后荧光细胞的频率,确定转染效率。列出了全部细胞中以%计的gfp阳性细胞相对数目。所有测定的平均转染效率是在30%-40%之间。‘hrm’:高分辨率熔点pcr阳性细胞定义为,与野生型细胞相比,展示明确趋异(双相)解链曲线样式的细胞。‘k.o.’表示不携带有功能的dph1或dph2基因拷贝并且因此抵抗dt的细胞的频率。‘int.‘表示携带pac表达盒并因此抵抗pm的细胞的频率。‘a-d’表示独立(四次重复)实验的各样品。
实施例3
检测crispr/cas9介导的dph1和dph2基因杂合敲除
仅使得dph1或dph2的一个等位基因修饰的细胞无毒素抗性。为了鉴定并定量这些事件,高分辨率熔解(hrm)pcr按照如stahl等人描述的相似方式应用(stahl等人,2015):转染后24小时,通过facs(facsaria,bdbiosciences)将单细胞沉积在96孔板中并且培养直至汇合。用pbs洗涤细胞并通过添加每孔40μl细胞裂解缓冲液(roche)裂解。在平板摇床(titramax1000,heidolph)上按750转/分钟室温温育15分钟后,细胞裂解物用pcr级h2o作1:5稀释。5μl细胞裂解物与高分辨率熔解(hrm)主混合物(roche)并与覆盖grna靶序列的引物混合。在lc480ii(roche)中根据生产商的方案进行pcr和hrm。通过与mcf7-wt细胞相比,改变的解链曲线鉴定靶基因已编辑的克隆。
借助细胞活力测定法,具有双相解链曲线的细胞仍可能拥有一个野生型等位基因或可能具有两个等位基因失活。因此,将这类克隆扩增(无毒素或嘌呤霉素选择)并进行细胞活力分析以区分杂合(毒素敏感)和纯合(抗性)敲除细胞。这些测定法在含有10,000个细胞的平底96孔平板中于37℃在增湿的5%co2条件下进行。接种后24小时,细胞暴露于毒素72小时。存活细胞的代谢活性由根据生产商说明书进行的
实施例4
检测盒整合事件
用于定向整合的crispr/cas模块含有无启动子以避免瞬时表达的嘌呤霉素(pm)抗性基因表达盒。因此,通过确定细胞对pm的敏感性,检测重组序列向基因组的整合情况。施加浓度500ng/μl的pm(定量消除野生型mcf7细胞)以选择稳定整合表达盒的mcf7细胞。采用如上文对dt选择所述的相同程序,选择pm抗性集落,并且按照如上文对毒素抗性集落所述的相同方式可视化和定量(在选择开始后12天和14天之间)。采用非靶向导序列(乱序grna,图1)获得的pm抗性集落的数目反映非特异性整合背景。采用靶基因grna获得的抵抗pm但是毒素敏感的集落增加(高于非特异性背景),反映整合入仅一个靶等位基因,另一个dph1或dph2等位基因保持不受影响。也具有dt抗性的pm抗性集落具有表达盒整合入至少一个靶等位基因中,而另一等位基因失活或失活并伴有另一整合事件。这些分析的结果汇总于表1中。
实施例5
白喉毒素抗性测定法和hrm-pcr的组合区分和定量纯合和杂合dph1基因失活事件
白喉毒素(dt)对真核翻译延伸因子2(eef2)上的白喉酰胺进行adp-核糖基化并且因而使eef2失活。这不可逆地停止蛋白质合成并杀死细胞。白喉酰胺是确定的组氨酸,其借助多个白喉酰胺合成基因置于eef2上,所述基因包括白喉酰胺生物合成1基因=dph1。彻底失活mcf7细胞中的全部dph1等位基因(例如,通过基因编辑)阻止毒素靶白喉酰胺的合成。这使得细胞抵抗假单胞菌外毒素a(pe)和dt(stahl等人)。结果,失活dph1的全部有功能拷贝产生‘dt抗性’表型(图3)。可以通过如实施例2-4中所述方式计数毒素抗性集落,以快速和稳健方式检测该表型的频率(结果在上表和图3中)。
因为存在仅一个剩余有功能的dph1等位基因对毒素敏感性而言即足够(图1和图2),所以抗性表型特异地定义了具有全部dph1等位基因敲除的细胞。可以通过对培养细胞直接进行的hrm-pcr测定法(实施例3中描述),鉴定仅一个等位基因修饰的细胞。与野生型片段相比,对crispr/cas靶位点的修饰改变dph1-基因来源的pcr片段的解链温度。这由hrm谱图中的双相解链曲线反映(4图中示例性显示)。由于两个等位基因上crispr-cas9介导的基因失活事件罕见相同,因此具有彻底基因失活的许多细胞也将显示双相hrm曲线。这些细胞可以依据它们的毒素抗性表型而区别于单等位基因改变。
因此,细胞上高通量hrm-pcr测定法和毒素选择(集落计数)测定法的组合能够实现对杂合和纯合dph1基因特异性修饰事件的定量。
实施例6
嘌呤霉素抗性测定法检测并区分基因特异性和非特异性整合事件的频率
嘌呤霉素-n-乙酰基-转移酶(pac),其由所应用的crispr/cas9质粒的整合盒编码,使pm失活并且因此使细胞抵抗pm(vara等人,1986)。因此,可以按照上文对dt抗性集落所述的相同方式,通过pm抗性测定法检测并定量pac表达盒的整合(施加pm替代dt作为选择剂,图3)。与仅因特异的和纯合的靶基因失活产生的dt抗性相反,pm抗性标记出任何整合事件,与整合位置无关。可以通过将暴露于靶基因特异性向导rna的pm抗性细胞的数目与暴露于乱序非特异性向导的细胞的数目比较,解析位点特异性整合与非特异性整合的频率(参见图3)。
实施例7
比较crispr-cas9所致dph1基因失活事件和定向整合事件
为了比较靶特异性失活、整合和非靶整合的频率,将编码dph1特异性crispr-cas9模块的质粒(实施例1,图1)转染入mcf7细胞。这些细胞随后接受hrm-pcr以及集落计数测定法以检测dt抗性和pm抗性。图5中总结这些测定法的结果,并且上表1中可获得各个数据集。
图5a显示dph1基因彻底失活,表明全部dph1等位基因的功能丧失以全部已转染细胞的约6%的频率发生(全部细胞的2.5%视为40%的转染效率,表1)。dph1基因失活显示出对匹配性向导rna序列的绝对依赖性:乱序向导不产生任何dt依赖性集落。图5b中显示dph1基因上hrm'命中'与dt抗性集落出现情况的比较。这些分析揭示,单等位基因失活(毒素敏感hrm命中)的发生频率两倍于双等位基因失活(dt抗性细胞)。
图6显示pm抗性集落的频率和dt抗性集落频率和pm抗性集落频率的比较。这些结果表明,dph1双等位基因失活以高于介导pm抗性的pac表达盒整合30-50倍的效率发生(图6a,不区分pac整合的位置或合子性)。与施加dph1特异性向导rna相比,乱序向导(在相同条件下)产生少2倍的pm抗性集落。这表示crispr/cas9/dph1-向导介导的pac基因整合偏好地发生在dph1基因处,但是没有绝对特异性。根据dph1靶部位处的偏好性整合,采用dph1向导所获得的许多pm抗性集落有毒素抗性(图6b)。采用乱序向导所获得的pm抗性集落均无dt抗性。
这些结果显示,crispr/cas9介导的靶基因失活(甚至同时在两个等位基因上失活)高度特异性发生并且频率比定向整合高得多(图6c)。定向整合不仅发生频率更低,而且对于向导rna限定的位置,特异性更小。
实施例8
crispr/cas9基因编辑的分析/定量方案的适用性和结果可以转移至其他靶基因
本文呈现的结果(包括观察到定向基因失活和整合之间的大差异)是crispr-cas9介导的基因编辑的一般特征或dph1基因的特别之处?为了解决这个问题,相同的实验方案应用于crispr-cas9诱导的dph2基因修饰。dph2位于不同于dph1的染色体上,具有不同的序列并且编码不同的酶。但是,dph2也对白喉酰胺合成为必需并且dph2缺乏使细胞按照与dph1缺乏相同的方式抵抗dt(stahl等人)。因而,为鉴定并区分dph1调节作用而在本文中报道和开发的检测和定量原则也可以适用于分析crispr/cas9介导的dph2改变。
图7中列示了dph2基因编辑的结果,随后是dt抗性和pm抗性评估结果:与对dph1的观察相符,观察到纯合dph2基因失活事件的频率高于pac表达盒整合约40倍。dph2失活严格依赖于存在关连向导rna,而盒整合具有位点偏好性,但是对靶基因没有绝对的特异性(比较采用乱序向导rna情况下dph2向导的频率)。dph1基因和dph2基因编辑结果的惊人相似性(即便两者是不同染色体上的不同基因)清楚地说明,用此分析系统获得的知识和规则可转移至其他合适的基因,并且为以下提供了合理基础:本文中报道的方法具有广泛适用性。
实施例9
应用如本文所报道的方法:比较和优化基因操作模块
dph1和dph2基因编辑实验的结果高度可比,即便两者是不同染色体上的不同基因。因此可以合理地推断:用此分析系统获得的'规则'和知识可以推广至其他基因。因此,如本文所报道的方法可以用于解析基因修饰模块组成的总体影响的分析中。
因此,如本文所报道的方法已经用于比较和优化基因操作模块。图8显示了如何评估不同长度的crispr/cas向导rna的基因失活以及整合的效率和特异性并就其基因失活和/或整合的效率和特异性进行比较。所用施加的向导rna均靶向dph1内部的相同序列段,但是长度从14至26个碱基不等(图1详细描述向导rna)。记录dt抗性集落的频率以反映靶基因特异性纯合基因失活。同时,评估pm抗性集落数目和dt-pm双重抗性集落数目以监测盒整合情况。
如预期,向导rna长度影响基因失活效率,20mer赋予最大的dph1基因失活效率。缩短互补段至18或16个碱基或使其延长至26个碱基保留明显的特异性基因失活功能,但效率低于20mer。缩减向导rna片段至小于16个碱基(14mer)降低dph1失活功能至检测水平以下。
整合效率(通过计数pm-抗性事件评估,比较并参见图5b)也受向导rna长度影响。小于16mer的向导(14mer)仅产生少数pm抗性集落,不超过乱序对照背景水平。对16mer,18mer,20mer,22mer,24mer和26mer观察到明显高于背景的定向整合,用16-18mer实现最佳插入效率。采用更大的寡聚核酸未实现效率增益,实际上,更大的尺寸(包括20mer)显著减少了特异性插入事件。
已经发现,整合事件(pm-r)和失活事件(dt-r)之间的比率可以算作‘特异性指示物’以鉴定基因失活事件最少情况下发生特异性整合的条件。如果想要定向整合且不造成太多无效靶基因损伤,则这类条件可能有利。
低数值(例如,pm抗性集落相对于dt-r集落而言少)反映整合相对于同时发生的失活事件而言无效。高数值(pm抗性集落更多和/或dt-r集落的数目相对减少)反映在crispr/cas影响的靶基因处的整合更高效。
图8b显示依赖于向导rna长度的计算的特异性系数。已经观察到,对于16-18mer,‘插入/失活’值最高,并且对于含有20个或更多碱基的向导rna,存在明显下降。这表明,20mer对定向基因失活而言相当高效(与先前观察一致,详细描述向导rna),然而较短的向导看起来对特异性整合而言更高效。较短的向导不仅改善整合效率(pm抗性集落的数目总体更高),还改善有效基因编辑和无效基因编辑之间的比率(无插入的dt-r集落减少)。
实施例10
比较不同基因编辑方案的效率和特异性
为了比较rna指导的cas9的不同变体以及zfn介导的基因编辑的基因失活和整合事件、效率和特异性,grna长度和组成保持恒定(20mer,靶向dph1)。作为变量,应用三种不同的编辑酶:
(i)‘spcas9’特指来自化脓性链球菌(streptococcuspyogenes)的cas9核酸酶,该酶可以视为当前标准应用(ran等人,2013,fu,sander等人,2014);
(ii)spcas9-hf1是非特异性dna结合作用降低、脱靶活性降低并且因此被提出具有更高精确度及特异性的spcas9工程化变体(kleinstiver,pattanayak等人,2016);
(iii)zfn介导的基因编辑模块,其借助设计的锌指基序(即,蛋白质-核酸相互作用,与核酸杂交相反)识别靶序列(miller,holmes等人,2007;urnov,rebar等人,2010)。
按照与先前实施例中对grna长度分析描述的相同方式,记录dt抗性集落的频率以反映靶基因特异性纯合基因失活。评估pm抗性集落和dt-pm双重抗性集落以评估监测盒整合情况。
比较基因失活和盒整合的总体效力,使用crispr/spcas9观察到最高的失活以及整合值。对于crispr/spcas9-hf,与crispr/spcas9相比,定向基因失活事件较少,低于dt抗性集落数目的20%。pm抗性集落(整合)数目和dt-pm双重抗性集落(盒整合和定向同时基因失活)数目也强烈地下降。在相同条件下,应用zfn模块导致dt抗性集落(纯合基因破坏)数目减少到小于使用crispr/spcas9观察到的事件的60%。因此,与spcas9相比,定向基因失活效率低大约2倍,而与工程化的spcas9-hf1相比,增加大约2-3倍。pm-抗性集落(整合)数目并未在crispr/spcas9和zfn之间显著差异。与crispr/spcas9相比,zfn的dt-pm双重抗性集落(盒整合同时伴基因失活)或多或少地(30%)减少。
计算dt抗性(靶基因失活)集落/dt+pm双重抗性(靶位点整合)集落比率(从公开得到总体效率)显示,crispr/spcas9及crispr/cas9-hf以及zfn系统产生相同水平(约4x10-3)的定向整合事件/一个纯合/彻底基因失活事件。因此,尽管采用现有编辑系统时基因修饰总体效率是变动的,但高于靶基因失活背景的盒整合‘特异性’被确定为对评价的全部三个方案并无显著差异。
因此,如本文所报道的方法可以因而用于评价其他编辑系统或支持编辑事件的添加物以鉴定改进的编辑模块和/或条件。
表:用不同基因编辑实体转染的mcf-7细胞的集落计数和表型频率
用编码不同基因组编辑系统(spcas9,spcas9,zfn)的质粒转染mcf-7细胞。如上文所述应用spcas9构建体。根据kleinstiver,pattanayak等人,2016,spcas9-hf包含n497a/r661a/q695a/q926a置换。同时,grna由scrna并行替换以解析非特异性活性。dph1特异性zfn从sigmaaldrich
实施例11
dna修复调节物对基因编辑效率和特异性的影响
如本文所报道的方法(包括集落测定法以在dph基因编辑后确定dt抗性细胞和pm抗性细胞)也可以用来解析调节dna修复机制的化合物的影响。例如,已经描述非同源末端接合(nhej)过程的抑制物调节基因编辑事件(ma,y等人,2016)。按照相同的方式,同源重组(hr)的激活物可以增加定向盒整合的效率(song,j等人,2016)。
为了评价并定量dna修复调节物对基因编辑效率和特异性的影响,将crispr/spcas9和靶向dph1的20mergrna与调节dna修复机制的化合物组合。转染基因编辑模块前4小时或转染后18小时,施加抑制物scr7直至转染后72小时以抑制dna连接酶iv。按照相同的方式,添加rad51调节物rs-1(刺激rad51的化合物1)以刺激hr。两种化合物按照不影响mcf7细胞生长或活力的剂量施加:scr7为1μm和rs-1为8μm,并且当两者组合时1μm+8μm(scr7+rs1)。随后记录dt抗性集落、pm抗性集落和双重抗性集落的频率以反映这些不同条件下靶基因失活事件和盒整合事件的效率和特异性。
可以显示,通过转染前4小时直至转染后72小时添加scr7,对nhej的调节没有对dt抗性集落的数目(反映靶基因失活频率)产生可检测的影响。还可以显示,与对照转染相比,转染前4小时共施加scr7和rs-1以及转染后18小时至72小时施加scr7导致dt抗性集落的数目小幅(仍明显)降低。因此,可以显示,dna连接酶iv介导的nhej可以影响crispr/spcas9所致的靶基因失活到某种程度。尽管其对dt-抗性集落总体数目的影响有限,但可以显示转染前4小时直至转染后72小时施加scr7显著地刺激(大约2倍,下表)pm抗性集落的频率。这提示,scr7增加盒整合事件,从而证实当施用scr7时增强有效基因编辑的先前观察(may等人,2016)。因此,这些结果提供证据:如本文所报道的方法可以用来确定这类影响。
可以显示,通过转染前4小时直至转染后72小时添加hr刺激物rs-1对重组的调节,增加了pm抗性集落的数目大约2倍,同时不影响采用乱序向导获得的集落的数目(下表)。还可以显示,当启动编辑后18小时至72小时施加rs1时,rs1不影响盒整合的效率。因此,这些结果提供证据:如本文所报道的方法可以用来确定这类影响。
可以显示,基因编辑中连接酶iv(nhej)抑制作用和hr刺激作用的组合(通过转染前4小时直至转染后72小时添加scr7以及rs-1)减少了dt抗性细胞的出现并且进一步提高pm抗性细胞的频率(下表)。可以显示,与所有其他方案相比,这种方案还导致了最高的pm抗性集落/dt抗性集落比率。因此,这些结果提供证据:如本文所报道的方法可以用来确定这类影响。
可以从这些实验得出结论,如本文所报道的方法可以用来确定化合物、化合物组合、化合物施加时间的影响以及影响基因编辑方案结果的潜在机制。因此,如本文所报道的方法因此可用于鉴定可以增强期望的基因编辑效果及减少不需要的‘副作用’的化合物或条件。
表:基因编辑期间暴露于scr7和/或rs-1的mcf-7细胞的集落计数和表型频率
如前文所述,将mcf-7细胞以用于spcas9介导的编辑的质粒转染。接种相等数目的细胞(接种的细胞数目)并在启动编辑后72小时用dt或pm处理。
(a)转染前4小时添加scr7(1μm终浓度)、rs-1(8μm终浓度)或scr7+rs1(分别为1um+8um)。
(b)rs1和scr7施加时间(转染前4小时vs.转染后18小时)对化合物调节的spcas9-grna介导的基因编辑的影响。(*)差异显著,p<0.008。
a
b
实施例12
鉴定和定量zfn介导的dph1基因编辑
使得dph1的全部染色体拷贝均失活的mcf7细胞有dt抗性。因此,当zfn所致基因失活和/或盒整合时dtr集落的出现和频率提供了失活全部基因拷贝的效力的量度。zfn识别序列(caggtgatggcggcgctggtcgtatccggggcagcggagcag,剪切位点,seqidno:10)来自nm_001383.3(dph1-wt)并且获自sigmaaldrich。用于此位置的pac整合盒获自origene。将mcf7细胞如上文所述用(i)gfp表达质粒、(ii)编码靶向dph1的zfn的质粒和(iii)靶向dph1的pac整合盒转染。在确定转染效率后,将细胞接种在6孔板中。为了定量纯合敲除事件(dtr),接种20,000个细胞,并且为了定量整合事件(pmr)或双重抗性,接种40,000个细胞。接种后3天,将rpmi交换成含有dt或pm或两者的rpmi培养基。每2-3天更换培养基。在开始毒素暴露后第12天和第14天之间,将细胞用pbs洗涤3次并随后用冰冷的亚甲基蓝(50%etoh中的0.2%)染色,随后流水下洗涤并且显微镜下用5mm网格箔计数集落数目。
实施例13
定量hr调节物和nhej调节物对crispr/cas9介导的编辑的影响
基因编辑期间施加刺激rad51的化合物1(rs-1)以调节同源重组(hr)。rs-1(sigmaaldrich)溶解于dmso中以产生10mg/ml母液,后者紧临施加至细胞之前稀释于rpmi培养基中。存活力(promegactg)测定法鉴定rs-1终浓度8μm是对mcf7不造成生长抑制作用或毒性作用的剂量(存活力:1μm-100%;3.7μm–100%;11μm–97%;33μm-61%)。
基因编辑期间施加dna连接酶iv抑制物scr7以调节非同源末端接合(nhej)。scr7(sigmaaldrich)溶解于dmso中以产生10mg/ml母液,后者紧临施加至细胞之前稀释于rpmi培养基中。存活力(promegactg)测定法鉴定终浓度1μm是对mcf7不造成生长抑制作用或毒性作用的剂量(存活力:0.37μm–100%;1.1μm-100%;3.3μm–97%;10μm-88%)。
在‘早期暴露′设置下,转染基因编辑模块前4小时,添加scr7(1μm终浓度)或rs-1(8μm终浓度)或scr7+rs-1(1μm+8μm终浓度)至mcf7细胞。对于‘晚期暴露’,转染后18小时添加scr7(8μm终浓度)或rs-1(1μm终浓度)至mcf7细胞。在两种情况下,细胞暴露于调节物直至转染后96小时,即‘早期暴露’由处理总计100小时组成,‘晚期暴露’由处理总计78小时组成。
在其上确定dna修复调节物的影响的系统由具有dph120mergrna的crispr/spcas9模块组成,其中将所述模块如先前实施例中所述那样转染入mcf7细胞并且接受后续dt选择和pm选择。记录dtr集落、pmr集落和双重抗性集落的频率以反映基因失活事件和盒整合事件。
实施例14
统计结果
进行非配对、双尾studentt检验以在两种处理之间作单一比较。通过单因素方差分析,随后tukey真实显著性差异(hds)事后检验,统计地分析多重比较。显著差异由p-值<0.05定义。图中由student’st检验确定的显著性水平由对应于p<0.05、p<0.01和p<0.001的一个、二或三个星号指示。同样,由tukey’shds检验确定的显著性水平由λ、φ或ψ指示。
- 还没有人留言评论。精彩留言会获得点赞!