靶分子的串联条形码添加以便以单实体分辨率对靶分子进行绝对定量的制作方法
本发明涉及用于在乳液液滴内以高通量方式标记来自实体例如细胞的核酸和其它生物分子,同时保持单个实体信息的完整性的方法和系统。
背景技术:
由于考虑细胞群的异质性可能对理解不同生物系统的功能和行为具有重要性的卓见,单细胞分析越来越受欢迎。克隆单细胞生物群或多细胞生物群的单个细胞,即使同一组织或细胞类型的细胞之间,通常表现出不同的基因调节或蛋白质表达模式。因此日益认识到,分析大量细胞群的传统分析方法得到的是整体意见,尽管这些意见具有信息性,但仅反映主要的生物学机制并且不能鉴别细胞间的变化。
rna水平被认为是表型异质性的有用标志物,因此,进行了大量工作来分析单细胞中的rna含量。包括荧光原位杂交(fish)或报告基因与荧光蛋白融合的探针依赖性方法被不依赖探针的rna-seq技术取代,在rna-seq技术中,细胞rna分子被转化为cdna,接着使用下一代测序技术并行测序。单细胞rna-seq需要分离单个细胞,将细胞rna转化为cdna以及cdna文库的大规模并行测序。
微流体装置的快速扩展引起了基于阀的微流体芯片的发展,在所述微流体芯片中,细胞被隔离在纳升反应室中(streets等人,2014)。然而,这种方法仍受限制,这不仅由于成本原因,而且还因为目前用所述芯片处理的单细胞的数量仍少于100个/运行。或者,微流体液滴还提供可隔离细胞的区室。通常,一相的液滴通过利用微流体两相流中的毛细管不稳定性在另一个不混溶相中产生。向任一相或两相中添加表面活性剂使液滴稳定以防止聚结并使其可用作离散微反应器。
此外,因为单细胞rna-seq分析可能需要分析数千甚至数百万代表性的单独的细胞,所以已经开发了条形码添加策略来降低测序成本并增加通量。使用唯一细胞鉴定符,可以汇集大量的细胞来同时测序,因为随后可以通过唯一细胞条形码将每个读数分配到其来源细胞(islam等人,2012)。
组合条形码策略和将细胞区室化到液滴中时的主要技术挑战是确保每个液滴携带不同的条形码,从而保持单细胞信息的完整性。
已经描述了几种方法来解决这个问题。可以将每个细胞与添加有独特条形码的颗粒如珠粒(macosko等人,2015)或水凝胶微球体(klein等人,2015)共包封在纳升级液滴中。这些颗粒中的每一个含有超过108个单独的引物,所述引物具有相同的“细胞条形码”。在这种方法中,为了确保一个液滴仅包含一个颗粒,所产生的液滴的数量大大超过注入的颗粒或细胞的数量,使得一个液滴通常含有零个或一个细胞和零个或一个颗粒(macosko等人,2015;klein等人,2015)。
或者,可以使用由96小滴制造器组成的微流体装置产生条形码文库乳液,该微流体装置产生数百万个小滴,这些小滴含有高浓度的96个条形码中的单个条形码(rotem等人,2015)。接着将每个带细胞小滴与一个条形码小滴配对并融合。然而,为了确保每个带细胞小滴与至多一个条形码小滴融合,实际上只有一半的带细胞小滴与条形码小滴融合。此外,两个带细胞小滴与单个条形码小滴融合或两个条形码小滴与单个带细胞小滴融合的情况在所得标记中引入误差并且是潜在的噪声来源(rotem等人,2015)。
因此,无论使用何种方法确保每个液滴携带不同的条形码,这都会导致液滴占有率受限,液滴的有用部分减少,从而导致通量减少。
此外,即使rna水平已被认为是表型异质性的有用标志物,但当前方法仅提供有限的信息,因为蛋白质或其它生物分子的水平不能用相同的系统评估。实际上,迄今为止,单细胞水平下的蛋白质表达的定量通常基于荧光成像方法,所述定量对于表型状态的完全表征至关重要。
因此,非常需要使得可在单实体水平下高通量定量rna、蛋白质和其它目标生物分子的新方法和装置。
技术实现要素:
本发明提供一种以高通量方式标记来自多个实体的任何靶分子,同时保持单个实体信息的完整性的新方法。
因此,在第一方面,本发明涉及一种标记来自多个实体的多个分子靶,同时保持单个实体信息的完整性的方法,所述方法包含
提供包含含有标记分子靶的液滴的第一组乳液液滴,其中这些液滴的每一个含有源自不超过一个实体的多个分子靶,并且其中在这些液滴的每一个中,用分子鉴定dna序列标记每个分子靶,所述分子鉴定dna序列包含(i)唯一分子鉴定(umi)条形码,所述唯一分子鉴定(umi)条形码对于每个分子靶是不同的,和(ii)突出端或产生突出端的限制性位点;
提供包含含有实体鉴定序列的液滴的第二组乳液液滴,其中这些液滴的每一个含有至少一个实体鉴定序列,所述实体鉴定序列是包含唯一实体鉴定(uei)条形码和产生突出端的限制性位点的dna序列,优选双链dna序列,所述唯一实体鉴定(uei)条形码对于第二组的每个液滴是不同的;
将第一组的液滴与第二组的液滴融合,其中将第一组的一个液滴与第二组的不超过一个液滴融合;和
任选地在限制酶消化之后,将uei条形码与标记分子靶连接,以及
任选地破坏所述乳液。
所述方法还可以包含
将多个实体包封在乳液液滴内,每个液滴含有不超过一个实体,并且任选地在液滴内裂解所述实体以释放分子靶;
用探针标记所述分子靶,每个探针包含
捕获部分,其能够与分子靶或与所述分子靶连接的衔接子特异性结合或连接,和
dna部分,其包含(i)邻近捕获部分并且包含唯一分子鉴定(umi)序列的区域和(ii)远离捕获部分并且包含突出端或产生突出端的限制性位点的区域,
从而获得第一组乳液液滴。
所述方法还可以包含
(i)将多个实体鉴定序列和扩增反应混合物一起包封在乳液液滴内,每个液滴含有不超过一个实体鉴定序列,和
在液滴内扩增实体鉴定序列
从而获得第二组乳液液滴,或
(ii)在uei校准物存在下将多个实体鉴定序列包封在乳液液滴内,其中至少一些液滴包含一个或数个实体鉴定序列和一个或数个uei校准物,并且其中所述uei校准物是包含唯一校准物条形码和一个或两个产生突出端的限制性位点的dna序列,优选双链dna序列,所述唯一校准物条形码对于每个uei校准物和每个液滴是不同的;和
在液滴内扩增实体鉴定序列和/或uei校准物;
从而获得第二组乳液液滴。
优选地,在融合第一组和第二组的液滴之后,通过限制酶消化和连接(i)uei校准物和实体鉴定序列的相容突出端以及(ii)实体鉴定序列和标记分子靶的相容突出端来组装(i)uei校准物条形码和uei条形码以及(ii)uei条形码和标记分子靶。
或者,在融合第一组和第二组的液滴之后,可以通过限制酶消化和连接(i)uei校准物和标记分子靶的相容突出端以及(ii)uei校准物和实体鉴定序列的相容突出端或i)uei校准物和实体鉴定序列的相容突出端以及(ii)实体鉴定序列和标记分子靶的相容突出端来组装(i)uei校准物条形码、uei条形码和标记分子靶。
或者,可以在扩增之前通过实体鉴定序列和uei校准物的相容突出端来组装实体鉴定序列和uei校准物,并且在融合第一组和第二组的液滴之后,通过i)uei校准物和标记分子靶的相容突出端或(ii)实体鉴定序列和标记分子靶的相容突出端将包含uei校准物和uei条形码的扩增片段连接于标记分子靶。
优选地,至少一些分子靶是核酸并且至少一些探针包含
捕获部分,其是通过常规的watson-crick碱基配对相互作用驱动核酸分子靶的特异性识别的单链dna区域,和
dna部分,其包括包含所述唯一分子鉴定(umi)序列的3'单链区域和包含所述突出端或产生突出端的限制性位点的5'双链区域。
优选地,使用所述探针标记所述核酸分子靶作为用于dna聚合酶合成分子靶的互补链的引发位点。
在具体实施方式中,至少一些分子靶是rna分子并且所述dna聚合酶是逆转录酶。
在一些实施方式中,至少一些探针包含捕获部分,所述捕获部分是
(i)与分子靶特异性结合并且与dna部分直接结合的结合部分,
(ii)包含与分子靶特异性结合的第一结构域和与dna部分结合的第二结构域的嵌合蛋白,或
(iii)与分子靶和蛋白桥特异性结合的结合部分,所述蛋白桥包含与结合部分结合的第一结构域和与dna部分结合的第二结构域。
优选地,(i)结合部分或嵌合蛋白的第一结构域选自与分子靶或一类分子靶特异性反应的抗体、配体/抗配体对的配体、肽适体、核酸适体、蛋白质标签或化学探针(例如自杀底物),优选抗体,(ii)蛋白桥的第一结构域是结合免疫球蛋白的细菌蛋白,优选蛋白a的结构域a至e,和/或(iii)蛋白桥或嵌合蛋白的第二结构域选自snap-标签、clip-标签或halo-标签,优选snap-标签。
在一些优选实施方式中,至少一些探针包括包含特异于分子靶的抗体部分和蛋白桥的捕获部分,所述蛋白桥包含与抗体部分的fc区域结合的第一结构域和与dna部分结合的第二结构域,优选snap-标签。
本发明还涉及一种以单实体分辨率定量来自多个实体的一个或数个分子靶的方法,所述方法包含
根据本发明方法标记所述分子靶;
捕获所述标记分子靶,
扩增包含umi、uei条形码和任选的uei校准物条形码的序列,
对扩增序列进行测序。
对umi、uei条形码和任选的uei校准物条形码的测序使得可明确地将每个分子靶分配到液滴/实体,因此定量每个液滴/实体中分子靶的含量。
优选地,所述实体是在其外表面上暴露分子靶的细胞或颗粒或水包油乳液液滴。更优选地,所述实体是细胞。
当实体是在其外表面上暴露分子靶的颗粒或乳液液滴时,该方法还可以包含
-用本文所定义的的探针标记分子靶,和
-将与标记分子靶连接的实体包封在乳液液滴内,每个液滴含有不超过一个实体,
从而获得第一组乳液液滴。
本发明还涉及试剂盒用于根据本发明方法标记来自多个实体的多个分子靶或用于根据本发明方法以单实体分辨率定量来自多个实体的一个或数个分子靶的用途,其中所述试剂盒包含
微流体装置,其包含
-第一乳液再注入模块或芯片上液滴产生模块;
-第二乳液再注入模块或芯片上液滴产生模块
-液滴配对模块,和
-耦合液滴融合与注入的模块,
其中乳液再注入模块和/或芯片上液滴产生模块与液滴配对模块流体连通并且在液滴配对模块的上游,液滴配对模块与耦合液滴融合与注入的模块流体连通并且在耦合液滴融合与注入的模块的上游,
和任选的,
-一个或数个本文所定义的探针;和/或
-一个或数个本文所定义的实体鉴定序列;和/或
-一个或数个本文所定义的uei校准物;和/或
-一个或数个适合于扩增实体鉴定序列和/或uei校准物的引物;和/或
-水相和/或油相;和/或
-提供此种试剂盒的使用指南的活页。
或者,本发明涉及试剂盒用于根据本发明方法标记来自多个实体的多个分子靶或用于根据本发明方法以单实体分辨率定量来自多个实体的一个或数个分子靶的用途,其中所述试剂盒包含
-一个或数个本文所定义的探针;和/或
-一个或数个本文所定义的实体鉴定序列;和/或
-一个或数个本文所定义的uei校准物;和/或
-一个或数个适合于扩增实体鉴定序列和/或uei校准物的引物;和/或
-水相和/或油相;和/或
-微流体装置,优选地本发明的微流体装置,和任选的
-提供此种试剂盒的使用指南的活页。
附图说明
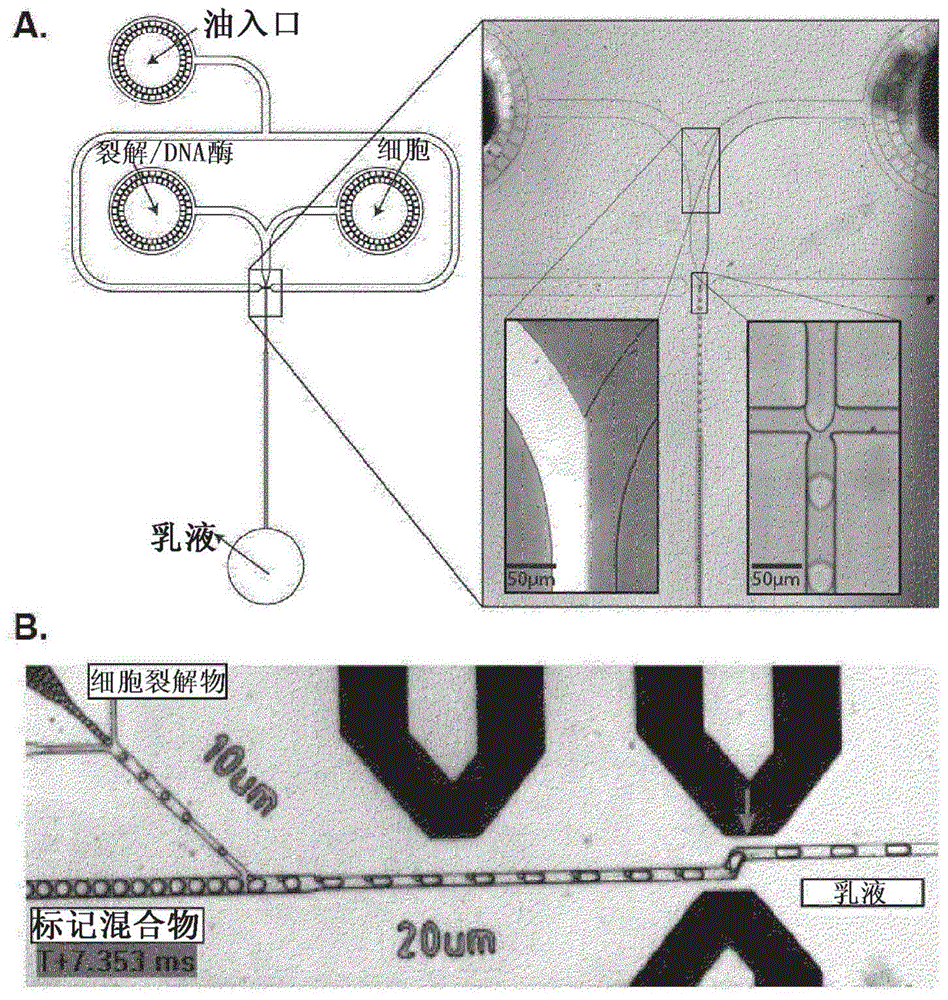
图1:用于单细胞分子标记的基于液滴的微流体平台。a.单细胞个体化和裂解。将含有细胞的水流与含有裂解剂和任选的双链特异性dna酶的水溶液流组合。产生乳液,收集并且温育以使得细胞裂解和dna降解可发生。b.液滴融合。将含有细胞裂解物的液滴再注入液滴融合微流体芯片中,并与芯片上产生的含有标记混合物(逆转录混合物、dna探针、抗体……)的液滴同步。接着将液滴对在通过电极对之间时在融合点(箭号)处融合。
图2:用于靶向标记核酸的基于dna的合成探针的示例性实施方式。在该实例中,探针的序列被设计用于定量金黄色葡萄球菌(staphylococcusaureus)的rnaiii。
图3:dna探针的示例性实施方式,其包含驱动特异性识别与rna分子靶连接的dna衔接子的捕获部分。前腺苷酸化的衔接子(5'-app寡核苷酸)用作t4连接酶的底物,并因此与rna分子连接。所述探针的捕获部分然后与dna衔接子特异性杂交。
图4:dna探针的示例性实施方式,其包含捕获部分,该捕获部分是在其5'末端包含5',5'-腺嘌呤基焦磷酰基部分(app)的5'单链dna区。此部分用作t4连接酶的底物,并因此与rna分子连接。
图5:嵌合探针的示例性实施方式。a.基于适体的探针。该探针由rna或dna适体组成,该rna或dna适体特异于靶分子并与合成的dna标记部分融合。b.包含捕获部分的嵌合探针,该捕获部分包含蛋白桥(snap-标签和蛋白a)和特异于分子靶的抗体。c.捕获部分的示意性组织。
图6:实体鉴定序列的示例性实施方式。实体鉴定序列包含恒定区、唯一限制性位点、uei条形码和测序引物退火序列。
图7:实体鉴定序列(uei)的多重包封:结果和使用uei校准物校正。使用包封在同一区室中的2个uei和2个校准物的简化情况来举例说明两种情形。a.没有校准物的情况下uei的多重包封。将两个细胞与2个不同uei一起包封在不同的液滴中。uei添加之后,来自同一细胞的靶分子与2个不同uei结合。这将导致靶分子被错误分配至仅代表细胞的子级分的群集,并且单细胞信息的完整性将受到损害。b.在校准物存在下uei的多重包封。将两个细胞与2个不同uei和2个不同校准物一起包封在不同的液滴中。uei添加之后,来自同一细胞的靶分子和校准物都与2个不同uei结合。在该过程结束时,第一分析定义了与同一校准物组相结合的uei的群集。接着将用属于同一群集的uei标记的靶分子簇集在一起,并保持了单细胞信息的完整性。
图8:uei校准物的示例性实施方式包含两个产生突出端的限制性位点(rs),第一限制性位点产生与包含uei条形码的经消化的实体鉴定序列的突出端相容的突出端,并且第二限制性位点产生与标记分子靶的突出端相容的突出端。在该实施方式中,用于向标记分子靶添加uei的消化/连接步骤导致形成包含标记分子靶、uei条形码和uei校准物条形码的三部分分子。
图9:uei校准物的示例性实施方式。该分子显示为pcr扩增产物(双链dna)。uei校准物包含测序引物退火序列、间隔子、uei校准物条形码(uei校准物)、唯一限制性位点和恒定区,所述恒定区包含使得可扩增uei校准物的引物结合位点。
图10:适合于实施本发明方法的微流体装置的示例性实施方式。将第一组和第二组乳液液滴再注入装置中(左侧显微照片),用油流间隔开并在两个再注入通道的接点处同步。同步的液滴可循环进入长通道中,该通道的宽度大于小液滴但比大液滴窄。因此,小液滴捕捉大液滴,并且在通道的出口处形成液滴对(中间的显微照片)。最后,液滴对处于注入点处,在该注入点处输注酶溶液,并且电极的存在引发液滴融合和酶递送两者(右侧显微照片)。
图11:同向流动液滴发生器。指示了微流体装置的关键尺寸。通道的深度是10μm。
图12:液滴荧光分析仪。指示了微流体装置的关键尺寸。深度是15μm。荧光测量点用开放箭头指示。
图13:含有完整或裂解细菌的橙色标记液滴的荧光图。上图:含有完整细菌的液滴的荧光图。每个橙色峰都对应于一个液滴。每个橙色峰中观察到的绿色尖峰对应于荧光颗粒,因此是完整细菌。下图:含有裂解细菌的液滴的荧光图。每个橙色峰对应于一个液滴。此外,具有与橙色峰相同宽度的均匀绿色(例如从左边开始第二个峰)的存在指示荧光标记的核酸已被释放到液滴中,因此细菌已经被裂解。
图14.油包水液滴的明视野和绿色荧光成像。在
图15:唯一鉴定符(ui)制备中的主要步骤。
图16:pcr扩增和(共)扩增uei校准物和uei的分析。左图:pcr扩增uei(泳道1)、uei校准物(泳道2)或两者一起(泳道3)的分析。右图:在本体中(泳道4)和在液滴中(泳道5)pcr共扩增uei校准物和uei的分析。uei校准物的预期大小和uei扩增产物大小的位置分别用开放箭头和闭合箭头标记。泳道l对应于低范围序列梯(sm1203,fermentas)。两种凝胶均为8%天然聚丙烯酰胺-tbe1x凝胶。
图17:液滴发生器。指示了微流体装置的关键尺寸并且通道深度是40μm。
图18.液滴微微注射器(picoinjector)。指示了关键尺寸并且通道深度是40μm。接地电极和正电极分别以浅灰色和深灰色显示。
图19.dna标记效率的分析。上图:带有uei校准物的dna(黑色方块)与带有uei的dna(虚线方块)重组之后在限制性位点(空心方块)处正确形成ui使引物6和10的退火位点在同一dna上,从而使得可进行qpcr。中图:对于以两个uei校准物/uei比率在本体中和在乳液中进行的实验给出ct值。此外,在限制/连接酶存在(+)或不存在(-)下进行标记反应。最后,对于每个反应,给出分析凝胶上相应泳道的数字。左下图:8%天然聚丙烯酰胺-tbe1x上qpcr产物的分析。泳道l对应于低范围序列梯(sm1203,fermentas)。用黑色箭头指示预期大小的产物的位置。右下图:索引化文库的凝胶纯化。在tbe中的1%天然琼脂糖凝胶上纯化索引化ui的文库。泳道l对应于1kb序列梯(sm1163,fermentas)。用黑色箭头指示预期大小的产物的位置。白色虚线框显示用于测序的回收的条带。
图20.用于分析测序数据的生物信息学算法。
图21:液滴中的条形码分布和标志出现次数。左图:uei校准物和uei在液滴中的分布。针对uei校准物(灰色虚线柱)以及uei(空心柱)显示了每个液滴的不同条形码序列的数量的分布。右图:在标志中进行ui簇集后,确定了序列库中标志的出现次数。
图22.各种条形码λ值下ui形成的分析。左图:ui形成的qpcr分析。带有uei校准物的dna(黑色方块)与带有uei的dna(虚线方块)重组之后在限制性位点(空心方块)处正确形成ui使引物6和10的退火位点在同一dna上,从而使得可进行qpcr。针对测试的不同λ(每个液滴的不同uei校准物和uei数量)给出ct值。右图:8%天然聚丙烯酰胺-tbe1x上qpcr产物的分析。泳道l对应于低范围序列梯(sm1203,fermentas)。黑色箭头指示了预期大小的产物的位置。
图23.uei校准物和uei在液滴中的分布图。有意去除值1和2下的出现次数,因为它们含有显著的测序噪声。
图24:所用靶dna的方案表示。
图25:ui形成和接枝到靶dna的分析。左图:ui形成和接枝到靶dna的qpcr分析。带有uei校准物的dna(黑色方块)与带有uei的dna(虚线方块)重组之后在限制性位点(空心方块)处正确形成ui以及在模拟消化产物的靶dna(圆点方块)末端处连接ui使引物6和3的退火位点在同一dna上,从而使得可进行qpcr。表格中给出ct值。右图:8%天然聚丙烯酰胺-tbe1x上qpcr产物的分析。泳道l对应于低范围序列梯(sm1203,fermentas)。黑色箭头指示了预期大小的产物的位置。
图26.液滴中ui制备和靶dna标记的示意性工作流程。
图27.条形码添加芯片。指示了关键尺寸并且通道深度是40μm。接地电极和正电极分别以浅灰色和深灰色显示。
图28.靶dna-ui索引化文库的纯化凝胶。在tbe中的1%天然琼脂糖凝胶上纯化索引化文库。泳道l对应于1kb序列梯(sm1163,fermentas)。黑色箭头指示了预期大小的产物的位置。白色虚线框显示用于测序的回收的条带。
图29.用于对测序数据去卷积并确定每个液滴/细胞的靶分子的拷贝数的生物信息学算法。
图30.液滴与液滴的可靠性。对于每个液滴呈现每个液滴中鉴定的dna分子的数量。请注意,具有低dna含量(1.8至0.5之间)的液滴可能是噪音,但将其保留在分析中以不使其产生偏见。
图31.用于逆转录rna-iii的引物的方案表示。
图32.2pl液滴发生器。指示了装置的关键尺寸。通道的深度是10μm。
图33.微流体液滴融合仪。指示了关键尺寸并且通道深度是15μm。接地电极和正电极分别以浅灰色和深灰色显示。
图34.逆转录产物的分析。上图:逆转录之后,使rt引物延伸并含有引物21的退火位点。产生的cdna含有两种引物结合位点(20和21),并且可以通过qpcr检测。表格中给出ct值。下图:8%天然聚丙烯酰胺-tbe1x上qpcr产物的分析。左图、中图和右图的凝胶分别对应于以每个液滴1000、100和10个rna开始的实验。泳道1、4和7是阴性对照,泳道2、5和8对应于在本体中进行的实验,并且泳道3、6和9对应于在液滴中进行的反应。泳道l对应于低范围序列梯(sm1203,fermentas)。黑色箭头指示预期大小的产物的位置,而空心箭头显示小的寄生性副产物。
图35.1%琼脂糖凝胶-tbe上pcr产物的分析。最初用不含随机区域的模板(泳道1)、n2x5模板(泳道2)、n4443模板(泳道3)和n15模板(泳道4)制备ui。黑色箭头指示了预期大小的产物的位置。黑色竖直条显示短的寄生性副产物。
图36.制备nabab-dna/igg复合物的工作流程。
图37.nabab-dna/igg复合物的制备。左图:温育具有nabab蛋白(泳道2)和不具有nabab蛋白(泳道1)的bg标记的荧光dna。l指示分子量梯。右图:将nabab蛋白单独温育(泳道1),与bg标记的荧光dna一起温育(泳道2)以及与增加浓度的igg一起温育(泳道3为62.5μg/ml,泳道4为112μg/ml,泳道5为225μg/ml)。温育之后,将反应产物加载到天然聚丙烯酰胺凝胶上,并且在不进行另外染色的情况下通过成像凝胶荧光(由与dna缀合的alexa488发光)显示dna分子的位置。
图38.ui与nabab-dna复合物的连接。左图:ui接枝到dna标记的蛋白质的qpcr分析。带有uei校准物的dna(黑色方块)与带有uei的dna(虚线方块)重组之后在限制性位点(空心方块)处正确形成ui以及在与蛋白质共价连接的dna的末端通过相容限制性位点连接ui使引物6和10的退火位点在同一dna上,从而使得可进行qpcr。表格中给出ct值。右图:1%琼脂糖凝胶-tbe1x上qpcr产物的分析。泳道l对应于低范围序列梯(sm1203,fermentas)。黑色箭头指示了预期大小的产物的位置。
具体实施方式
发明人构思了一种以高通量方式标记来自多个实体的任何靶分子,同时保持单个实体信息的完整性的新方法,所述高通量方式即允许每次运行分析数千个实体。该方法基于串联分子条形码添加,其中用以下标记所有分子靶(核酸、蛋白质……):(i)第一唯一条形码(唯一分子鉴定条形码或umi条形码),所述第一唯一条形码对于来自实体的每个分子靶是不同的,和(ii)编码分子靶所来源的实体的标签序列,即唯一实体鉴定条形码或uei条形码,所述标签序列对于每个实体是不同的,但对于源自同一实体的所有分子靶是相同的。一旦执行该串联条形码添加,就可以在下一代测序的单次运行中以单实体分辨率进行所有分子靶的绝对定量,从而使得该方法高度灵敏并且成本有效。
在第一方面,本发明涉及一种标记来自多个实体的多个分子靶的方法,所述方法包含
提供包含含有标记分子靶的液滴的第一组乳液液滴,其中这些液滴的每一个含有源自不超过一个实体的多个分子靶,并且其中在这些液滴的每一个中,用分子鉴定dna序列标记每个分子靶,所述分子鉴定dna序列包含(i)唯一分子鉴定(umi)条形码,所述唯一分子鉴定(umi)条形码对于每个分子靶是不同的,和(ii)突出端或产生突出端的限制性位点;
提供包含含有实体鉴定序列的液滴的第二组乳液液滴,其中这些液滴的每一个含有至少一个实体鉴定序列,所述实体鉴定序列是包含唯一实体鉴定(uei)条形码和产生突出端的限制性位点的dna序列,优选双链dna序列,所述唯一实体鉴定(uei)条形码对于第二组的每个液滴是不同的;
将第一组的液滴与第二组的液滴融合,其中将第一组的一个液滴与第二组的不超过一个液滴融合;和
任选地在限制酶消化之后,将uei条形码与标记分子靶连接。
可以使用本发明方法标记来自任何类型实体的分子靶。
如本文所用,术语“实体”是指在其表面上包含或暴露如下所定义的分子靶的任何实体。特别地,该术语是指在其外表面上暴露分子靶的细胞,或指在其外表面上暴露分子靶的颗粒或乳液液滴,优选水包油乳液液滴。
如本文所用,术语“细胞”是指原核细胞或真核细胞,如动物、植物、真菌或藻类细胞。待处理的细胞群可以是同质的,即仅包含一种细胞类型,或可以是异质的,即包含数种细胞类型。在一些实施方式中,细胞群获自组织样品,优选动物组织样品,更优选病理样品,如肿瘤样品。在一些其它实施方式中,细胞群是细菌、真菌或藻类细胞群,优选细菌或真菌细胞群。该群体可以包含相同种的细菌、真菌或藻类或不同种的细菌、真菌或藻类。
术语“颗粒”和“珠粒”在本文中可互换使用,并且是指尺寸为50nm至10μm的任何固体支持物,优选球形固体支持物,其适合于在其外表面上暴露一个或多个分子靶。特别地,这些术语可以指聚合物珠粒(例如聚丙烯酰胺、琼脂糖、聚苯乙烯)、乳胶珠粒、磁性珠粒或水凝胶珠粒。使分子靶如核酸或蛋白质与珠粒共价或非共价结合的方法是本领域技术人员所公知的,并且可购得各种技术。特别地,这种结合可以通过颗粒表面上的反应基团进行。例如,核酸可以通过碳二亚胺介导的5'-磷酸和5'-nh2修饰的核酸分别与氨基和羧基珠粒的末端连接与表面连接。蛋白质还可以通过任何合适的方法与珠粒共价或非共价连接,如使用硫酸盐、脒、羧基、羧基/硫酸盐或氯甲基修饰的珠粒。
术语“实体”还可以指在其外表面上暴露分子靶的乳液液滴,优选水包油乳液液滴。分子靶可以通过在液滴表面上暴露的可以与带有his标签的蛋白质特异性相互作用的反应基团如次氮基三乙酸酯或通过能够与目标分子靶共价或非共价相互作用的任何其它官能部分与液滴共价或非共价连接。技术人员可以使用任何已知方法来产生在其外表面上暴露分子靶的这种乳液液滴,特别是国际专利申请wo2017/174610中描述的方法。
本发明方法使得可在单次运行中标记来自大量实体的分子靶。因此,如本文所用,术语“多个实体”是指至少1,000个实体,优选至少5,000个实体,更优选至少10,000个实体,并且甚至更优选至少50,000个实体。
如本文所用,术语“靶分子”或“分子靶”是指任何种类的分子,并且特别可以是可能存在于细胞中的任何种类的分子。分子靶可以是生物分子,即活生物体中天然存在的分子,或活生物体中天然不存在的化合物,如药物、毒剂、重金属、污染物等……。优选地,分子靶是生物分子。生物分子的实例包括但不限于核酸例如dna或rna分子,蛋白质如抗体、酶或生长因子,脂质如脂肪酸、糖脂、固醇或甘油脂,维生素,激素,神经递质和碳水化合物例如单糖、低聚糖和多糖。术语“多肽”、“肽”和“蛋白质”可互换地用于指氨基酸残基的聚合物,并且不限于最小长度。蛋白质可以包含任何翻译后修饰,如磷酸化、乙酰化、酰胺化、甲基化、糖基化或脂化。如本文所用,术语“核酸”或“多核苷酸”是指任何长度的核苷酸的聚合形式,所述核苷酸是核糖核苷酸或脱氧核糖核苷酸。
本发明方法的主要优点之一是可以在单次运行中标记不同类型的分子靶,如蛋白质和核酸。因此,术语“多个分子靶”可以指相同分子的不同拷贝,例如,相同mrna或相同蛋白质的不同拷贝,或可以指不同分子的不同拷贝,例如mrna的不同拷贝和蛋白质的不同拷贝。
在一个实施方式中,分子靶是相同分子的不同拷贝。优选地,分子是生物分子,更优选核酸或蛋白质,甚至更优选rna分子或蛋白质。在另一个实施方式中,分子靶是不同分子的不同拷贝。优选地,所述分子是生物分子,更优选是核酸和/或蛋白质,甚至更优选rna分子和/或蛋白质。在一个具体实施方式中,分子靶是至少两种不同核酸、优选rna的不同拷贝。在另一个具体实施方式中,分子靶是至少两种不同蛋白质的不同拷贝。在另一个实施方式中,分子靶是一种或数种核酸、优选rna的不同拷贝,和一种或数种蛋白质的不同拷贝。
在优选实施方式中,使用一个或数个微流体系统来实施本发明方法,即使用微流体系统实施该方法的至少一个步骤。在一些实施方式中,使用数个微流体系统实施该方法,所述微流体系统例如产生第一组乳液液滴的微流体系统、产生第二组乳液液滴的微流体系统以及融合两组、并入uei条形码并且任选地进行一些后续步骤的微流体系统。在一些其它实施方式中,使用如下微流体系统实施该方法,其中产生第一组乳液液滴和/或第二组乳液液滴,并且其中将两组液滴融合。
如本文所用,术语“乳液液滴”、“液滴”和“微流体液滴”可互换使用,并且可以指油包水乳液液滴(也称为w/o液滴),即完全被油相包围的水相的隔离部分;水包油乳液液滴(也称为o/w液滴),即完全被水相包围的油相的隔离部分;水包油包水乳液液滴(也称为w/o/w液滴),其由油滴内的水滴,即水核和油壳组成,该油滴被水性载流体包围;或油包水包油乳液液滴(也称为o/w/o液滴),其由水滴内的油滴,即油核和水壳组成,该水滴被油性载流体包围。优选地,该术语是指w/o乳液液滴。
根据外部环境,液滴可以是球形或其它形状。通常,液滴的体积小于100nl,优选小于10nl,并且更优选小于1nl。例如,液滴的体积可以在2pl至1nl,优选2至500pl,更优选2至100pl范围内。优选地,液滴具有均匀的直径分布,即液滴可以具有一种直径分布,使得液滴的不超过约10%、约5%、约3%、约1%、约0.03%或约0.01%的平均直径大于液滴平均直径的约10%、约5%、约3%、约1%、约0.03%或约0.01%。优选地,乳液是单分散乳液,即包含相同体积液滴的乳液。用于产生这种均匀直径分布的技术是本领域技术人员公知的(参见例如wo2004/091763)。
水相通常是水或水性缓冲溶液,例如但不限于tris-hcl缓冲液、tris-乙酸盐缓冲液、磷酸盐缓冲盐水(pbs)或乙酸盐缓冲液。优选地,水相是水性缓冲溶液。任选地,水相可以包含牛血清白蛋白或添加剂,如pluronic。在优选实施方式中,选择水相以与在本发明的过程中进行的酶促反应相容,所述酶促反应如酶消化、扩增、连接等……。此种水相的实例包括但不限于cutsmart限制酶缓冲液(newenglandbiolabs)。
用于产生乳液液滴的油相可以选自氟化油,如fc40油
乳液液滴包含一种或数种表面活性剂。所述表面活性剂可以帮助控制或优化液滴尺寸、流动和均匀性以及稳定水性乳液。用于制备本发明中使用的乳液液滴的合适表面活性剂通常是非离子型的,并且含有至少一个亲水性头部和一个或数个亲脂性尾部,优选一个(二嵌段表面活性剂)或两个(三嵌段表面活性剂)亲脂性尾部。所述亲水性头部和尾部可以直接连接,或通过间隔子部分连接。合适的表面活性剂的实例包括但不限于基于脱水山梨糖醇的羧酸酯,如脱水山梨糖醇单月桂酸酯(span20)、脱水山梨糖醇单棕榈酸酯(span40)、脱水山梨糖醇单硬脂酸酯(span60)和脱水山梨糖醇单油酸酯(span80);聚乙二醇和聚丙二醇的嵌段共聚物,如三嵌段共聚物ea-表面活性剂(raindancetechnologies)、dmp(二吗啉代磷酸酯)表面活性剂(baret,kleinschmidt等人,2009)和jeffamine表面活性剂;聚合的基于硅的表面活性剂,如abilem90;tritonx-100;和氟化表面活性剂,如pfpe-peg和全氟化聚醚(例如krytox-peg、dupontkrytox157fsl、fsm和/或esh)。在本发明的上下文中,优选的表面活性剂是氟化表面活性剂,即氟表面活性剂。
在一些具体实施方式中,乳液液滴在其界面处包含一种或数种官能化表面活性剂。如本文所用,“官能化表面活性剂”是指在亲水性头部中的一个或亲脂性尾部上,优选在亲水性头部上带有至少一个官能部分的表面活性剂。如本文所用,“官能部分”实际上是为表面活性剂提供目标功能的任何化学或生物实体。例如,官能部分能够在表面活性剂和目标分子靶之间产生共价或非共价相互作用。由于使用此类官能化表面活性剂,分子靶可以暴露在乳液液滴的表面上。这些液滴的界面可以仅包含官能化表面活性剂或官能化和非官能化表面活性剂的混合物。官能化和非官能化表面活性剂之间的比率可以变化,并且可以容易地由技术人员调整。例如,官能化表面活性剂可以占总表面活性剂的1至100%(w/w),优选2至80%(w/w)并且更优选5至50%(w/w)。
优选地选择载体油中表面活性剂的总量以确保乳液的稳定性并且防止液滴自发聚结。通常,载体油包含0.5至10%(w/w),优选1至8%(w/w)并且更优选2至5%(w/w)表面活性剂。
乳液可以通过技术人员已知的任何方法制备。优选地,可以在微流体系统上制备乳液。
提供包含标记分子靶的第一组乳液液滴
第一组乳液液滴包含含有标记分子靶的液滴,其中这些液滴的每一个含有源自不超过一个实体的多个分子靶,并且其中,在这些液滴的每一个中,分子靶被分子鉴定dna序列标记。
根据实体的性质,第一组乳液液滴可以是w/o或o/w/o乳液。在一些实施方式中,实体是细胞或颗粒,并且第一组的乳液液滴是w/o乳液液滴。在一些其它实施方式中,实体是在其外表面上暴露分子靶的o/w乳液液滴,并且第一组的乳液液滴是o/w/o乳液液滴。
优选地,第一组乳液液滴包含至少10,000个液滴,优选至少100,000个液滴,优选至少500,000个液滴,甚至更优选至少1,000,000个液滴。
在一些实施方式中,第一组乳液液滴通过以下方法获得:
-将实体包封在乳液液滴内,每个液滴含有不超过一个实体,并且任选地在液滴内裂解所述实体以释放分子靶,并且
-标记所述分子靶。
在具体实施方式中,实体是在其外表面上暴露分子靶的颗粒或o/w乳液液滴,并且通过以下方法获得第一组乳液液滴:
-将实体包封在乳液液滴内,每个液滴含有不超过一个实体,并且
-标记所述分子靶。
在一些具体实施方式中,其中实体是在其外表面上暴露分子靶的颗粒或o/w乳液液滴,分子靶的标记可以在本体中进行,接着可以包封与标记分子靶连接的实体。
因此,当实体是在其外表面上暴露分子靶的颗粒或乳液液滴时,该方法可以包含
-标记所述分子靶,并且
-将与标记分子靶连接的实体包封在乳液液滴内,每个液滴含有不超过一个实体,
从而获得第一组乳液液滴。
在优选实施方式中,实体是细胞并且第一组乳液液滴通过以下方法获得:
-将实体包封在乳液液滴内,每个液滴含有不超过一个实体,
-在液滴内裂解所述实体以释放分子靶,并且
-标记所述分子靶。
为了确保单实体的分辨率,必须将实体从条形码添加过程的开始到结束进行限制和隔离。将实体包封到微流体液滴中是分隔所述实体的便利方式,并且特别适合于高通量方案。
本领域普通技术人员知道用于将细胞或颗粒包封在微流体液滴内的技术(参见例如国际专利申请wo2004/091763,该申请通过引用并入本文中)。如果细胞是粘附的或来自组织,则可以首先将其解离并且任选地过滤或离心以在包封之前去除两个或更多个细胞的团块。通常可以将细胞悬浮在水性缓冲液如pbs缓冲液中。
用于制备单分散w/o/w双乳液,即用于包封o/w液滴的方法也是技术人员所公知的,并且产生此类乳液的微流体系统是可商购的。
在包封期间,必须调整实体数量密度(每单位体积的实体)以最小化在同一液滴中捕获两个或更多个实体的发生率。特别地,可以以每个液滴少于1个实体的密度,优选每个液滴少于0.2个实体的密度包封实体,以防止共包封两个或更多个实体。在优选实施方式中,调整实体数量密度和平均占有率以确保大多数,优选至少98%或所有液滴中仅存在零个或一个实体。
通过使用任何公知的微流体方法将实体包封到微流体液滴中,可以以高频率制备带实体液滴,例如在0.5khz至15khz,优选1khz至10khz,更优选1khz至5khz范围内的高频率。
在实体是细胞的实施方式中,在包封之后,可以在液滴内裂解细胞以释放分子靶。可以使用技术人员已知的任何方法如使用物理、化学或生物学手段进行细胞裂解。特别地,可以使用辐射(例如uv、x或γ射线)或激光裂解细胞(参见例如rau等人,2004)。还可以通过渗透压休克或通过添加洗涤剂或酶来诱导裂解(参见例如kintses等人,2012;novak等人,2011;brown和audet,2008)。还可以通过热休克诱导裂解。
在一些实施方式中,由裂解剂诱导裂解。优选地,裂解剂包含一种或数种改变渗透平衡的组分、一种或数种洗涤剂和/或一种或数种酶。更优选地,裂解剂是tritonx-100、
在一个实施方式中,在包封之前将裂解剂直接添加到液滴的水相中。在此种实施方式中,可以在即将产生液滴之前,将含有细胞的水流与含有裂解剂的水溶液流组合(参见例如图1a)。接着可以产生乳液,收集并且温育以使得细胞裂解。
在另一个实施方式中,在液滴产生之后,通过任何已知技术如微微注射(picoinjection)或液滴融合将裂解剂引入液滴内。接着可以收集乳液并且温育以使得细胞裂解。
通常,将乳液在4℃至25℃范围内的温度下温育5分钟至1小时以使得细胞裂解。
或者,可以通过热处理诱导裂解。通常,在这种情况下,可以将乳液在高达95℃的温度下温育5分钟至1小时以使得细胞裂解。
技术人员可以容易地根据所用方法调整裂解期间的温育温度。
在一些实施方式中,其中第一组液滴的w/o界面包含官能化表面活性剂,通过细胞裂解释放的一些或所有分子靶可以被所述表面活性剂结合并且浓缩在液滴的内部w/o界面上。可以如国际专利申请wo2017/174610中所呈现,使用液滴反转将这些w/o液滴转化为o/w液滴。
在一些实施方式中,其中实体是在其外表面上暴露分子靶的颗粒或o/w乳液液滴,分子靶可以通过切割剂(例如限制酶)的作用从所述颗粒或从所述o/w乳液液滴的表面释放。
任选地,根据分子靶的性质,可以在收集和温育第一组乳液之前将一种或数种其它试剂添加到水相中。此类其它试剂的实例可以包括但不限于dna酶、rna酶、蛋白酶、蛋白酶抑制剂和/或核酸酶抑制剂。
在一些实施方式中,分子靶是rna,并且将一种或数种其它试剂,优选包含一种或数种dna酶和/或一种或数种蛋白酶的其他试剂添加到水相中。在一些其它实施方式中,分子靶是蛋白质和/或rna,并且将其它试剂,优选包含一种或数种dna酶的其他试剂添加到水相中。
可以将其它试剂和裂解剂同时添加到水相中,即在即将包封之前直接添加到液滴的水相中,或在液滴产生之后通过任何已知技术如微微注射或液滴融合添加。或者,可以依序添加其它试剂和裂解剂。特别地,可以在包封之前通过共流动含有实体的水溶液流和含有裂解剂的溶液流将裂解剂添加到水相中,并且可以在包封之后添加其它试剂,并且反之亦然,或者可以在包封之后依序添加裂解剂和其它试剂,例如单独的微微注射或液滴融合。
在优选实施方式中,将其它试剂和裂解剂同时加入到水相中,即在即将包封之前直接添加到液滴的水相中(参见例如图1a)。
在如上文所详述包封和任选地细胞裂解和/或消除/降解一些非靶向分子之后,用分子鉴定dna序列标记分子靶,所述分子鉴定dna序列包含(i)唯一分子鉴定(umi)条形码,所述唯一分子鉴定(umi)条形码对于每个分子靶是不同的,和(ii)突出端或产生突出端的限制性位点。
“umi序列”或“umi条形码”是将唯一条形码分配到每个分子靶的随机化核苷酸序列,因此使得可进一步进行最初存在于实体中或实体上的分子靶的数字检测/计数、其绝对定量和扩增偏差的校正。实际上,在液滴中,每个探针携带唯一鉴定号(umi),因此计数不同umi的数量给出了标记分子靶的绝对数量。优选地,umi序列是随机化核苷酸序列,其长度为至少5个核苷酸,优选长度为5至15个核苷酸,更优选长度为5至10个核苷酸。随机化序列可以是连续随机化核苷酸片段或半随机化核苷酸片段(即由恒定核苷酸间隔开的连续随机化核苷酸)。半随机化核苷酸片段的典型实例是数个随机化二核苷酸由恒定的二核苷酸间隔开的片段,或数个随机化三核苷酸由恒定的三核苷酸间隔开的片段。优选地,umi序列是半随机化核苷酸片段,特别是数个随机化二核苷酸由恒定的二核苷酸间隔开的片段。
需要突出端以如下文所详述,在与第二组的液滴融合之后添加uei条形码和任选的uei校准物条形码。该突出端可以是3'突出端或5'突出端,优选是3'突出端。
分子鉴定dna序列可以包含与限制酶产生的粘性末端相容的突出端(3'或5'突出端),或者可以包含产生突出端的限制性位点。
在一些实施方式中,分子鉴定dna序列包含与限制酶产生的粘性末端相容的突出端,优选地3'突出端。这种构造的选择确保了聚合酶的填充活性不会合成互补链,使得末端将保持用于后续uei添加的能力。另外,该突出端与用于消化双链dna序列的限制酶的切割产物相容,所述双链dna序列带有uei条形码或uei校准物并且如下文所详述包含在第二组的液滴中。通过使用消化和连接,该突出端使得可向每个umi序列添加uei和任选的uei校准物。
在一些其它实施方式中,分子鉴定dna序列包含产生突出端的限制性位点,即产生3'或5'突出端,优选3'突出端的限制性位点,所述突出端与用于消化双链dna序列的限制酶的切割产物相容,所述双链dna序列带有uei条形码或uei校准物并且包含在第二组的液滴中。在一个具体实施方式中,分子鉴定dna序列上产生突出端的限制性位点由与带有uei校准物的双链dna序列上产生突出端的限制性位点相同的酶识别(参见下文)。特别地,在融合第一组和第二组的液滴之后,可以在消化带有uei条形码的dna序列和带有uei校准物的dna序列的同时产生标记分子靶上的突出端。
或者,在实体是在其外表面上暴露分子靶的颗粒或o/w乳液液滴的实施方式中,分子靶可以通过限制酶的作用从所述颗粒或从所述o/w乳液液滴的表面释放,从而产生3'或5'突出端。任选地,分子鉴定dna序列还可以包含类型鉴定符序列,该序列是短预定义序列,优选具有4至8个核苷酸的长度,编码分子靶的性质(例如核酸或蛋白质)和/或身份(例如gfpmrna)。优选地,类型鉴定符序列邻近umi序列,更优选地直接与umi序列相邻。
在优选实施方式中,使用探针进行用umi条形码标记分子靶,每个探针包含
捕获部分,其能够与分子靶特异性结合或与分子靶或与所述分子靶连接的衔接子特异性连接,和
包含分子鉴定dna序列的dna部分,即(i)邻近捕获部分并且包含唯一分子鉴定(umi)序列和任选的类型鉴定符序列的区域,和(ii)远离捕获部分并且包含突出端,优选与限制酶产生的粘性末端相容的突出端或产生突出端的限制性位点的区域。优选地,远离捕获部分的区域包含3'突出端或产生3'突出端的限制性位点。
在远离捕获部分的区域包含3'突出端的实施方式中,5'末端是磷酸化的5'末端。
优选地,限制性位点或突出端与umi序列分隔至少10个核苷酸,优选至少20个核苷酸,并且更优选20至40个核苷酸。优选地,该分隔区域具有至少50℃,更优选至少55℃的熔解温度,富含gc以形成稳定的双链体,与细胞所来源的生物体中存在的核酸不具有任何序列同一性并且不含一个后续用于标记的限制性位点。在优选实施方式中,该区域对于所有探针是相同的。
在一个具体实施方式中,dna部分从捕获部分到限制性位点或突出端包含4至8个核苷酸、优选4个核苷酸的类型鉴定符序列,5至15个核苷酸、优选8个核苷酸的umi序列和包含限制性位点或突出端的20至40个核苷酸的区域。
捕获部分的性质和dna部分的结构可以根据分子靶的类型和/或化学结构而不同。因为可以同时标记不同类型和/或化学结构的分子靶,所以同一液滴中的探针可以具有不同结构。
在一些实施方式中,至少一些分子靶是核酸,并且至少一些特异于所述核酸的探针是包含以下的dna探针:
捕获部分,其是通过常规的watson-crick碱基配对相互作用驱动核酸分子靶的特异性识别或与所述分子靶连接的核酸衔接子的特异性识别的单链dna区域,和
dna部分,其包含(i)邻近捕获部分并且包含唯一分子鉴定(umi)序列和任选的类型鉴定符序列的3'单链区域,和(ii)远离捕获部分并且包含突出端,优选与限制酶产生的粘性末端相容的突出端或产生突出端的限制性位点的5'双链区域。
优选地,dna部分包含(i)邻近捕获部分并且包含唯一分子鉴定(umi)序列和任选的类型鉴定符序列的3'单链区域,和(ii)远离捕获部分并且包含3'突出端,优选与限制酶产生的粘性末端相容的3'突出端的5'双链区域。
这些dna探针可以通过技术人员已知的任何方法如化学合成来制备。
此种探针的示例性说明如图2所呈现。
在这些实施方式中,捕获部分的长度必须足以使得可通过杂交特异性识别靶分子。优选地,捕获部分是长度为至少8个核苷酸,优选长度为8至25个核苷酸,更优选长度为10至15个核苷酸的单链dna区域。
优选调整捕获部分与分子靶结合后形成的完美杂交体的熔解温度(tm)(例如通过调节特异于靶核酸的序列的长度)以使其在30℃至70℃范围内,优选在30℃至60℃范围内,更优选在40℃至60℃范围内,并且甚至更优选在40℃至50℃范围内。
优选地,特异于核酸分子靶的所有探针的熔解温度(tm)之间的差异低于3℃,更优选低于2℃,并且甚至更优选低于1℃。
捕获部分可以特异于特定的dna或rna,或者可以与所有rna共有的序列区域互补,例如捕获部分可以是poly-t区块,该区块与真核mrna的poly-a尾互补或与例如通过rna连接酶的作用向所有rna添加的核酸衔接子、优选dna衔接子互补。
因此,在一些实施方式中,捕获部分驱动与目标分子靶例如所有rna连接的核酸衔接子的特异性识别。这种衔接子可以是例如前腺苷酸化的寡核苷酸(5'-appoligo),其用作t4连接酶的底物,因此可以与任何rna分子连接。通常此种衔接子可以是长度为至少8个核苷酸,优选8至25个核苷酸,更优选长度为10至15个核苷酸的单链dna区域。此种实施方式的示例性说明如图3所呈现。
在一些其它实施方式中,至少一些分子靶是核酸,并且至少一些特异于所述核酸的探针是包含以下的dna探针:
捕获部分,其为能够与核酸分子靶连接的单链dna区域,和
dna部分,其包含(i)邻近捕获部分并且包含唯一分子鉴定(umi)序列和任选的类型鉴定符序列的5'单链区域,和(ii)远离捕获部分并且包含突出端、优选与限制酶产生的粘性末端相容的突出端或产生突出端的限制性位点的3'双链区域。
优选地,捕获部分是在其5'末端包含5',5'-腺嘌呤基焦磷酰基部分(app)的5'单链dna区。此种部分可以用作t4连接酶的底物,因此可以与任何rna分子的3'末端连接。在该实施方式中,捕获部分的5'app末端直接与rna分子靶相互作用,并且在t4连接酶存在下与所述靶标连接。此种实施方式的示例性说明如图4a和4b所呈现。
这些dna探针可以通过技术人员已知的任何方法如化学合成来制备。
优选地,捕获部分特异于特定的dna或rna分子,更优选地特异于特定的rna分子。
在一些实施方式中,可以使用上述探针,特别是所述探针的捕获部分标记核酸分子靶,作为用于dna聚合酶合成分子靶的互补链的引发位点。在逆转录或其它dna聚合反应之后,dna和/或rna分子靶被转化为添加有条形码的互补dna(cdna)。
在一些实施方式中,一些分子靶是rna分子并且dna聚合酶是逆转录酶。在探针通过其捕获部分与靶向rna或衔接子杂交之后,可以进行逆转录,并且可以合成互补dna(cdna)的第一条链,所述cdna包含探针的dna部分的umi序列。
应该认识到dna聚合反应或逆转录需要适当的条件,例如适当缓冲液和dna聚合酶的存在,适合于探针与靶向rna或dna退火的温度和酶的活性以及任选存在dtt。这些条件主要取决于聚合酶,并且可以根据供应商指南进行调整。
优选地,在这些实施方式中,所述方法还包含必要时在裂解步骤之后,
使分子靶与包含如上所述的dna探针、至少一种dna聚合酶、dntp混合物(datp、dctp、dgtp、dttp)和任选的适当缓冲液的标记混合物接触,和
使用聚合反应将rna分子靶转化为包含分子鉴定dna序列(umi)的cdna。
替选性地或附加性地,至少一些探针可以是由合成dna寡核苷酸即dna部分组成的嵌合分子,所述嵌合分子与第二分子即捕获部分共价结合,该第二分子特异性地且以高亲和力靶向靶分子并且使得可用可扩增且可读信号特异性标记任何分子。这些探针可以特异于任何类型的分子靶,优选特异于蛋白质分子靶。
这些探针的捕获部分可以是
(i)与分子靶特异性结合并且与dna部分直接结合的结合部分,
(ii)包含与分子靶特异性结合的第一结构域和与dna部分结合的第二结构域的嵌合蛋白,或
(iii)与分子靶和蛋白桥特异性结合的结合部分,所述蛋白桥包含与结合部分结合的第一结构域和与dna部分结合的第二结构域。
术语“特异性结合”在本文中用于指示该部分具有识别目标分子靶并与目标分子靶特异性相互作用的能力,同时与水相中存在的其它结构如可由其它探针识别的其它分子靶具有相对很少的可检测反应性。由于非共价力如静电力、氢键、vanderwaals力和疏水力,任何两个分子之间通常存在低亲和度,这不限于分子上的特定位点,并且在很大程度上与分子的身份无关。这种低亲和度可以引起非特异性结合。相比之下,当两个分子特异性结合时,亲和度远大于此类非特异性结合相互作用。在特异性结合中,每个分子上的特定位点相互作用,其中特定位点在结构上互补,从而形成非共价键的能力提高。可以通过使用例如biacore仪器进行结合或竞争性测定相对地确定特异性。分子x对其配偶体y的亲和力通常可以由解离常数(kd)表示。在优选实施方式中,代表捕获部分和目标分子靶之间亲和力的kd为1.10-7m或更低,优选1.10-8m或更低,并且甚至更优选1.10-9m或更低。
在一些实施方式中,至少一些探针包含捕获部分,该捕获部分是与分子靶特异性结合并且与dna部分直接结合的结合部分。优选地,结合部分与dna部分共价结合。
结合部分的实例包括但不限于抗体、配体/抗配体对的配体、肽和核酸适体、蛋白质标签或化学探针(例如自杀底物),其与分子靶或一类分子靶特异性反应。
配体/抗配体对的实例包括但不限于抗体/抗原或配体/受体。特别地,在一些实施方式中,分子靶是抗体,并且结合部分是被所述抗体识别的抗原,或反之亦然。在一些其它实施方式中,分子靶是受体,并且结合部分是被所述受体识别的配体,或反之亦然。
大量蛋白质标签是技术人员所公知的(参见例如young等人,biotechnol.j.2012,7,620-634)并且可以用于本发明。此类蛋白质标签的实例包括但不限于生物素(用于与抗生蛋白链菌素或抗生物素蛋白衍生物结合)、谷胱甘肽(用于与蛋白质或与谷胱甘肽-s-转移酶连接的其它物质结合)、凝集素(用于与糖部分结合)、c-myc标签、血凝素抗原(ha)标签、硫氧还蛋白标签、flag标签、polyarg标签、polyhis标签、strep-标签、ompa信号序列标签、钙调蛋白结合肽、几丁质结合结构域、纤维素结合结构域、s-标签和softag3等。
大量化学探针是技术人员所公知的(参见例如niphakis和cravatt,ann.rev.ofbiochem.2014,83,341-77和willems等人,bioconjugatechem.2014,25,1181-91)并且可以用于本发明。化学探针的实例包括但不限于亲电或光反应性基于活性的探针(abp),基于自杀底物的abp和基于抑制剂的abp。
在一个具体实施方式中,至少一些探针是基于适体的探针,即包含核酸或肽适体作为捕获部分的探针。与抗体类似,适体通过识别特定的三维结构与其靶标相互作用。适体可以特异性识别多种靶标,如蛋白质、核酸、离子或小分子,如药物和毒素。
肽适体由两端与蛋白质骨架如细菌蛋白硫氧还蛋白-a连接的短可变肽环组成。通常,可变环长度由十至二十个氨基酸组成。可以使用技术人员已知的任何方法,如酵母双杂交系统或噬菌体呈现(phagedisplay)来选择特异于目标靶标的肽适体。可以通过技术人员已知的任何方法制备肽适体,如化学合成或在重组细菌中产生,接着纯化。
优选地,至少一些探针包含核酸适体作为捕获部分。核酸适体是一类小核酸配体,其由rna或单链dna寡核苷酸组成,并且对其靶标具有高特异性和亲和力。用于产生特异于目标靶标的核酸适体的通过指数富集的配体系统进化(selex)技术是技术人员所公知的,并且可以用于获得特异于特定分子靶的适体。可以通过技术人员已知的任何方法制备核酸适体,如化学合成或用于rna适体的体外转录。优选地,用作捕获部分的核酸适体选自dna适体、rna适体、xna适体(包含异种核苷酸的核酸适体)和镜像异构体(spiegelmer)(其完全由非天然l-核糖核酸骨架组成)。此种探针的示例性说明如图5a所呈现。
在另一个具体实施方式中,至少一些探针是基于抗体的探针,即包含抗体作为捕获部分的探针。
本文的术语“抗体”以最广泛含义使用,并且特别涵盖单克隆抗体、多克隆抗体、抗体片段和其衍生物,只要其与目标分子靶特异性结合即可。特别地,抗体可以是全长单克隆或多克隆抗体,优选全长单克隆抗体。优选地,该术语是指具有含有fc区的重链的抗体。本文使用的“fc”、“fc片段”或“fc区”是指包含除第一恒定区免疫球蛋白结构域之外的抗体恒定区的多肽。因此,fc是指iga、igd和igg的最后两个恒定区免疫球蛋白结构域,ige和igm的最后三个恒定区免疫球蛋白结构域以及这些结构域n末端的柔性铰链。优选地,所述抗体是全长单克隆或多克隆igg抗体,优选全长单克隆igg抗体。目前市场上可获得大量特异性和高亲和力单克隆抗体。
如本文所用,术语“抗体片段”是指包含全长抗体的一部分,通常其抗原结合结构域或可变结构域的蛋白质。抗体片段的实例包括fab、fab'、f(ab)2、f(ab')2、f(ab)3、fv(通常是抗体单臂的vl和vh结构域)、单链fv(scfv)、dsfv、fd(通常是vh和ch1结构域)和dab(通常是vh结构域)片段、纳米抗体、小型抗体、双抗体、三抗体、四抗体、κ体、线性抗体和保留抗原结合功能的其它抗体片段(例如holliger和hudson,natbiotechnol.2005年9月;23(9):1126-36)。抗体片段可以通过各种技术制备,包括但不限于蛋白水解消化完整抗体以及重组宿主细胞(例如大肠杆菌或噬菌体)。这些技术是技术人员所公知的,并且在文献中有广泛的描述。优选地,抗体片段选自fab'、f(ab)2、f(ab')2、f(ab)3、fv、单链fv(scfv)片段和纳米抗体。
如本文所用,术语“抗体衍生物”是指本文提供的抗体,例如全长抗体或抗体片段,其中例如通过烷基化、peg化、酰化、酯或酰胺形成等化学修饰一个或多个氨基酸。特别地,该术语可以指本文提供的抗体,其被进一步被修饰以含有本领域已知且易于获得的其它非蛋白质性部分。
在一些实施方式中,捕获部分选自单克隆和多克隆抗体、fab'、f(ab)2、f(ab')2、f(ab)3、fv、单链fv(scfv)片段和纳米抗体以及其衍生物。在一些优选实施方式中,捕获部分选自单克隆抗体、scfv片段或纳米抗体。
在一些实施方式中,至少一些探针包含捕获部分,该捕获部分是包含与单个分子靶特异性结合的第一结构域和与单个dna部分结合的第二结构域的嵌合蛋白。优选地,所述第二结构域与所述dna部分共价结合。
嵌合蛋白的第一结构域与单个分子靶特异性结合。第一结构域的实例包括但不限于上述抗体、配体/抗配体对的配体、肽和核酸适体、蛋白质标签和化学探针。优选地,嵌合蛋白的第一结构域选自抗体和肽适体,更优选是单克隆抗体。
在一个具体实施方式中,嵌合蛋白的第一结构域是抗体,优选选自单克隆和多克隆抗体、fab'、f(ab)2、f(ab')2、f(ab)3、fv、单链fv(scfv)片段和纳米抗体以及其衍生物。更优选地,嵌合蛋白的第一结构域选自单克隆抗体、scfv片段或纳米抗体,并且甚至更优选选自scfv片段或纳米抗体。
与dna部分共价结合的第二结构域可以是使得可共价接枝单个核酸的任何结构域。此类结构域的实例包括但不限于snap-
可以使用任何公知的重组工程技术将用作捕获部分并且包含第一和第二结构域的嵌合蛋白制成融合蛋白形式,接着与探针的dna部分共价结合。
在一些实施方式中,至少一些探针包含捕获部分,该捕获部分包含(i)与分子靶特异性结合的结合部分,和(ii)蛋白桥,所述蛋白桥包含与结合部分结合的第一结构域和与dna部分结合的第二结构域。优选地,第二结构域与dna部分共价结合。第一结构域可以与结合部分共价或非共价结合,优选非共价结合。在第一结构域与结合部分非共价结合的实施方式中,优选通过交联第一结构域和结合部分而将非共价相互作用变为共价相互作用。
蛋白桥的第二结构域可以如上对于嵌合蛋白所述,即使得可共价接枝单个核酸的任何结构域。优选地,蛋白桥的第二结构域是snap-
蛋白桥的第一结构域可以是使得可与结合部分发生共价或非共价相互作用、优选非共价相互作用的任何结构域。此种结构域的实例包括但不限于结合免疫球蛋白的细菌蛋白,如蛋白a、蛋白a/g、蛋白g和蛋白l。
在一个实施方式中,蛋白桥的第一结构域是结合免疫球蛋白的细菌蛋白,并且结合部分是抗体,优选含有fc区的抗体,更优选全长单克隆或多克隆igg抗体,优选全长单克隆igg抗体。结合免疫球蛋白的细菌蛋白优选选自蛋白a、蛋白a/g、蛋白g和蛋白l、其一个或多个igg结合结构域和其功能衍生物。
蛋白a是金黄色葡萄球菌中存在的细胞表面蛋白。它具有结合哺乳动物抗体、特别是igg类抗体的fc区的性质。该蛋白质的氨基末端区域含有5个高度同源的igg结合结构域(称为e、d、a、b和c),并且羧基末端区域将蛋白质锚定在细胞壁和细胞膜上。蛋白a的所有五个igg结合结构域通过fc区与igg结合,并且原则上,这些结构域中的每一个足以与igg的fc部分结合。
因此,在一个具体实施方式中,蛋白桥的第一结构域选自蛋白a的结构域a、b、c、d和e、其组合以及其保留野生型蛋白a的igg结合功能的功能衍生物。优选地,蛋白桥的第一结构域包含蛋白a的结构域a至e。
可以使用任何公知的重组工程技术将蛋白桥制成融合蛋白,接着与探针的dna部分共价结合。
优选地,第一结构域位于蛋白桥的n末端部分,并且第二结构域位于蛋白桥的c末端部分。
任选地,蛋白桥还可以在c末端包含亲和标签(例如多组氨酸标签)以促进其纯化。
在一个具体实施方式中,蛋白桥包含结合免疫球蛋白的细菌蛋白,优选蛋白a的结构域a至e作为第一结构域,包含snap-
结合部分可以与蛋白桥共价或非共价结合。在一些实施方式中,蛋白桥可以与结合部分交联以确保长期的物理连接。
在上述嵌合探针中,dna部分由双链dna分子组成,所述双链dna分子包含突出端,优选与限制酶产生的粘性末端相容的突出端,或如上文对于dna探针所述的产生突出端的限制性位点。优选地,在dna部分中包含或由限制性位点产生的突出端是3'突出端。如上所述,dna部分包含唯一分子鉴定(umi)序列和任选的类型鉴定符序列。
优选地,在嵌合探针中,dna部分还包含邻近捕获部分的测序引物退火序列。标记分子靶之后,该序列使得可使用测序引物直接扩增。
优选地,在至少一些探针是嵌合探针的实施方式中,所述方法还包含必要时在裂解步骤之后使分子靶与包含至少一种嵌合探针的标记混合物接触。
可以在包封细胞之前(即通过直接包括在混合物中或通过共流)或在液滴产生之后通过任何已知技术如微微注射或液滴融合将包含探针和任选的进行dna聚合反应的试剂的标记混合物添加到液滴的水相中。优选地,在温育之后添加标记混合物,所述温育使得细胞裂解和/或可消除/降解一些非靶向分子。
在一个具体实施方式中,将标记混合物包封在w/o液滴中,并且将这些液滴与包含分子靶的液滴融合。包含标记混合物的每个液滴的含量应基本相同。制备此种乳液的方法以及产生、同步和融合乳液液滴的方法,特别是在微流体装置上,都是技术人员所公知的。
提供包含细胞鉴定序列的第二组乳液液滴
第二组乳液液滴包含含有实体鉴定序列的液滴,其中这些液滴的每一个含有至少一个实体鉴定序列。
实体鉴定序列是长度为40至100个核苷酸,优选长度为50至70个核苷酸的双链dna序列,所述实体鉴定序列包含唯一实体鉴定(uei)条形码,所述唯一实体鉴定(uei)条形码对于第二组的每个液滴是不同的,和产生突出端的限制性位点,优选产生3'突出端的限制性位点。
“uei序列”或“uei条形码”是将相同条形码分配到源自同一实体的每个分子靶的随机化核苷酸序列。优选地,umi序列是随机化核苷酸序列,其长度为至少8个核苷酸,优选长度为8至20个核苷酸,更优选长度为8至15个核苷酸。随机化序列可以是连续随机化核苷酸片段或半随机化核苷酸片段(即由恒定核苷酸间隔开的连续随机化核苷酸)。半随机化核苷酸片段的典型实例是其中数个随机化二核苷酸由恒定的二核苷酸间隔开的片段,或其中数个随机化三核苷酸由恒定的三核苷酸间隔开的片段。优选地,umi序列是半随机化核苷酸片段,特别是其中数个随机化二核苷酸由恒定的二核苷酸间隔开的片段。
包含在实体鉴定序列中的限制性位点可以在用相应的限制酶消化之后产生与标记分子靶的突出端相容的突出端,或与下述uei校准物的突出端相容的突出端,因此使得可向分子鉴定dna序列添加uei序列。优选地,该限制性位点是非回文切割位点。
任选地,实体鉴定序列还可以包含与uei条形码相邻并且位于限制性位点的相反末端的测序引物退火序列。
在优选实施方式中,实体鉴定序列包含使得可扩增带有uei的dna的恒定区,该恒定区与限制性位点相邻并且在uei条形码和存在时测序引物退火序列的相反末端。优选地,该恒定区的长度为15至35个核苷酸,优选20至30个核苷酸。优选地,恒定区具有在50℃至70℃之间的熔解温度。
此种实体鉴定序列的示例性说明如图6所呈现。
优选地,除了uei条形码序列之外,所有实体鉴定序列具有相同的序列,即它们表现出相同的测序引物退火序列,相同的限制性位点和相同的恒定区。
尽管对于每个液滴,uei序列必须是不同的,但它也必须以大量相同拷贝存在于每个液滴中。
因此,在一个实施方式中,所述方法还包含将多个实体鉴定序列与扩增反应混合物一起包封在乳液液滴内,其中每个液滴含有不超过一个实体鉴定序列,和在液滴内扩增实体鉴定序列,从而获得第二组乳液液滴。通过这种方法,可以在每个液滴中产生同一实体鉴定序列的超过一百万个拷贝,随后用于用相同且唯一的uei序列对先前标记的分子靶进行条形码添加。可以以单链或双链形式、优选以双链形式包封实体鉴定序列。
扩增反应混合物包含在液滴中进行dna扩增所需的所有试剂,即通常是dna聚合酶、引物、缓冲液、dntp、盐(例如mgcl2)等……。设计了引物以使得可完整扩增实体鉴定序列。在某些实施方式中,扩增依赖于交替的加热和冷却循环(即热循环)以实现连续的复制循环(例如pcr)。扩增划分在乳液液滴中的遗传元件的方法是技术人员所公知和广泛实践的(参见例如chang等人,labchip.2013年4月7日;13(7):1225-42;zanoli和spoto,biosensors2013,3,18-43)。特别地,可以通过任何已知技术进行扩增,所述技术如聚合酶链式反应(pcr)、基于核酸序列的扩增(nasba)、环介导的等温扩增(lamp)、解旋酶依赖性扩增(hda)、滚环扩增(rca)、多重置换扩增(mda)和重组酶聚合酶扩增(rpa)。本领域技术人员可以根据包封的遗传元件和分子靶的性质容易地选择合适的方法。优选地,通过pcr扩增将实体鉴定序列扩增到液滴中。
优选地,在该实施方式中,为了防止在一个液滴中共包封超过一个实体鉴定序列,从而保持単实体分辨率,将包含实体鉴定序列的dna溶液用例如扩增反应混合物强烈稀释,使得根据驱动分子分布的poisson统计,液滴占有率,即包含实体鉴定序列的液滴的百分比,限于少于20%,优选少于10%。因此,在具体实施方式中,所述方法还包含,在将实体鉴定序列和扩增反应混合物包封在乳液液滴中之前,稀释实体鉴定序列,优选用扩增反应混合物稀释,以获得少于20%、优选少于10%的液滴占有率。
为了提高液滴占有率,可以容许在一个液滴内包封数个实体鉴定序列。在这种情况下,发明人发现可以通过向实体鉴定序列添加uei校准物分子来保持单实体分辨率。因此,在一个优选实施方式中,方法还包含
在uei校准物和扩增反应混合物存在下将多个实体鉴定序列包封在乳液液滴中,其中至少一些液滴包含一个或数个实体鉴定序列和一个或数个uei校准物;和
使用多重反应在液滴内扩增实体鉴定序列和/或uei校准物,优选实体鉴定序列和uei校准物;
从而获得第二组乳液液滴。
可以以单链或双链形式、优选以双链形式包封实体鉴定序列和uei校准物。
优选地,对于多重反应,相对于特异于uei校准物的引物,使用较大量的特异于实体鉴定序列的引物。
将数个实体鉴定序列共包封在同一液滴中的可能性使得液滴占有率显著提高。实际上,调整实体鉴定序列溶液的稀释度以使得每个液滴共包封五个实体鉴定序列,使得液滴占有率接近100%。
优选地,调整实体鉴定序列溶液和uei校准物溶液的稀释度以使得每个液滴共包封2至10个实体鉴定序列,优选每个液滴4至6个实体鉴定序列和每个液滴2至10个uei校准物,优选每个液滴4至6个uei校准物。
uei校准物是dna序列,优选双链dna序列,其包含唯一校准物条形码,所述唯一校准物条形码对于每个uei校准物和每个液滴是不同的,和一个或两个产生突出端的限制性位点,所述产生突出端的限制性位点优选产生与经消化的实体鉴定序列和/或标记分子靶的突出端相容的突出端。
优选地,这些产生突出端的限制性位点是非回文切割位点。优选地,这些限制性位点产生3'突出端。优选地,这些限制性位点不同于实体鉴定序列上产生相容突出端的限制性位点和/或标记分子靶上产生相容突出端的限制性位点。“uei校准物序列”或“uei校准物条形码”是随机化核苷酸序列,其长度为至少15个核苷酸,优选长度为15至40个核苷酸,更优选长度为15至20个核苷酸。随机化序列可以是连续随机化核苷酸片段或半随机化核苷酸片段(即由恒定核苷酸间隔开的连续随机化核苷酸)。半随机化核苷酸片段的典型实例是其中数个随机化二核苷酸由恒定的二核苷酸间隔开的片段,或其中数个随机化三核苷酸由恒定的三核苷酸间隔开的片段。优选地,umi校准物条形码是半随机化核苷酸片段,特别是其中数个随机化二核苷酸由恒定的二核苷酸间隔开的片段。
在一个实施方式中,uei校准物包含一个产生突出端的限制性位点,该产生突出端的限制性位点在用相应限制酶消化之后,产生与经消化的包含uei条形码的实体鉴定序列的突出端相容的突出端。在该实施方式中,因为两种类型的分子,即uei校准物序列和实体鉴定序列,携带产生相容末端的限制性位点,所以用于向标记分子靶添加uei的后续消化/连接步骤使得在uei和uei校准物条形码之间形成不同的嵌合体。实际上,尽管大部分uei条形码将保持可用于对源自实体的标记分子靶进行条形码添加,但小部分uei条形码将与每个uei校准物条形码相结合。这些序列对构成每个液滴唯一的标志,并且使得可将每个被分析的分子再分配到其来源实体,尽管在一些液滴中存在多个实体鉴定序列(参见例如图7)。任选地,在该实施方式中,uei校准物还可以包含与uei校准物条形码相邻并且在产生与经消化的实体鉴定序列相容的突出端的限制性位点的相反末端的测序引物退火序列。任选地,uei校准物还可以在接近测序引物退火序列的末端包含使得可特异性捕获分子的结合标签。优选地,结合标签选自生物素或地高辛(digoxigenin),更优选是生物素。
优选地,在该实施方式中,uei校准物包含与限制性位点相邻并且在uei校准物条形码和存在时测序引物退火序列的相反末端的恒定区。该恒定区使得可扩增uei校准物,优选使用pcr扩增。特别地,该区域可以包含引物结合位点。优选地,该恒定区的长度为10至35个核苷酸,优选15至25个核苷酸。优选地,该恒定区与实体鉴定序列的恒定区正交以防止不期望的杂交。uei校准物还可以包含额外区域,该额外区域包含在uei校准物条形码和存在时测序引物退火序列之间。该区域用作测序引物退火序列和uei校准物条形码之间的间隔子,并且可以用于根据包含umi和uei条形码的扩增序列的长度调整包含uei和uei校准物条形码的扩增序列的长度以限制有效的扩增偏差。此种uei校准物的示例性说明如图9所呈现。
在另一个实施方式中,uei校准物包含两个产生突出端的限制性位点,第一限制性位点产生与经消化的包含uei条形码的实体鉴定序列的突出端相容的突出端,并且第二限制性位点产生与标记分子靶的突出端相容的突出端。在该实施方式中,用于向标记分子靶添加uei的后续消化/连接步骤使得可形成包含标记分子靶、uei序列和uei校准物条形码的三部分分子(参见例如图8)。
在该实施方式中,uei校准物可以包含与两个产生突出端的限制性位点中的每一个相邻的恒定区。这些恒定区使得可扩增uei校准物,优选使用pcr扩增。特别地,这些区域可以包含引物结合位点。优选地,这些恒定区的长度为10至35个核苷酸,优选15至25个核苷酸。优选地,这些恒定区与实体鉴定序列的恒定区正交以防止不期望的杂交。此种uei校准物的示例性说明如图9所呈现。
优选地,除了uei校准物条形码序列之外,所有uei校准物具有相同的序列,即它们表现出相同的限制性位点、相同的恒定区和任选的相同测序引物退火序列。
如上所述,可以使用技术人员已知的任何方法,优选多重pcr,在液滴内进行实体鉴定序列和uei校准物的多重扩增。扩增反应混合物包含在液滴中进行dna扩增所需的所有试剂,即通常是dna聚合酶、引物、缓冲液、dntp、盐(例如mgcl2)等……,并且设计引物以使得可完整扩增实体鉴定序列和uei校准物。
优选地,在uei和uei校准物条形码之间产生嵌合体的实施方式中,为了确保实体鉴定序列的大量过度呈现并使得在液滴融合之后对分子靶进行正确的条形码添加,相对于特异于uei校准物的引物,扩增反应混合物包含较大量的特异于实体鉴定序列的引物。更优选地,相对于特异于uei校准物的引物,扩增反应混合物包含更高至少5倍、优选至少10倍并且更优选至少100倍量的特异于细胞鉴定序列的引物。
在产生包含标记分子靶、uei序列和uei校准物条形码的三部分分子的实施方式中,扩增反应混合物可以包含类似量的特异于实体鉴定序列的引物和特异于uei校准物的引物。
任选地,可以在扩增之前通过uei序列和uei校准物的相容突出端来组装uei序列和uei校准物。在这种情况下,扩增反应因此直接扩增包含uei序列和uei校准物的片段。在与第一组液滴融合之后,所述片段被连接到标记分子靶上。
液滴融合并且向标记分子靶中并入uei条形码和任选的uei校准物条形码
如上所述,第一组乳液液滴包括用包含umi条形码的分子鉴定序列标记的分子靶,并且第二组乳液液滴包括包含uei条形码的实体鉴定序列和任选的uei校准物。
本发明方法包含将第一组的液滴与第二组的液滴融合,其中将一个第一组的液滴与不超过一个第二组的液滴融合。
可以使用技术人员已知的任何技术将第一液滴和第二液滴融合在一起产生组合液滴。例如,可以给予第一和第二液滴相反的电荷(即正电荷和负电荷,不一定具有相同大小),这可以提高两个液滴的电相互作用,使得可以由于其相反的电荷而发生液滴融合或聚结。例如,可以向液滴施加电场,可以使液滴通过电容器,化学反应可以使液滴带电等。
可以使用技术人员已知的任何技术确保一个第一组的液滴与不超过一个第二组的液滴融合。特别地,可以使液滴在到达聚结点之前通过配对通道配对。技术人员公知此种通道的使用以控制微流体液滴的融合。在使用配对通道的实施方式中,第一组和第二组的液滴应具有不同的尺寸。配对通道是长通道,其宽度大于最小液滴并且比最大液滴窄。结果,小液滴捕捉大液滴,并且在通道的出口处形成液滴对。该构造确保液滴在到达聚结点之前正确地配对。此种配对通道的一个示例性实施方式如图10中所说明。
在一些实施方式中,在单独的微流体系统上产生第一组的液滴和/或第二组的液滴并且将所述液滴再注入装置中。优选地,将液滴用油流隔开并在进入配对通道之前同步。
优选地,第一组的至少60%,更优选至少80%的液滴,甚至更优选至少95%与第二组的液滴融合。
在液滴融合之后,通过限制酶消化和连接将uei条形码并入标记分子靶中。
在第二组液滴不包含uei校准物的实施方式中,通过限制酶消化和连接标记分子靶和经消化的包含uei条形码的实体鉴定序列的相容突出端将uei条形码并入标记分子靶中。
在第二组的液滴包含uei校准物并且所述uei校准物包含一个产生突出端的限制性位点的实施方式中,可以通过限制酶消化和连接(i)uei校准物和实体鉴定序列的相容突出端以及(ii)实体鉴定序列和标记分子靶的相容突出端来组装(i)一方面uei校准物条形码和uei条形码以及(ii)另一方面uei条形码和标记分子靶。在该实施方式中,通过消化/连接耦合反应将实体鉴定序列附加到标记分子靶和uei校准物两者上。此种实施方式的示例性说明如图7b所呈现。或者,通过限制酶消化和连接i)uei校准物和实体鉴定序列的相容突出端以及(ii)实体鉴定序列和标记分子靶的相容突出端来组装uei校准物条形码、uei条形码和标记分子靶。
在第二组的液滴包含uei校准物并且所述uei校准物包含两个产生突出端的限制性位点的实施方式中,通过限制酶消化和连接(i)一方面uei校准物和标记分子靶的相容突出端以及(ii)另一方面uei校准物和实体鉴定序列的相容突出端来组装uei校准物条形码、uei条形码和标记分子靶。在该实施方式中,实体鉴定序列、uei校准物和标记分子靶一起形成三部分分子。此种实施方式的示例性说明如图8所呈现。
在一个实施方式中,包含在实体鉴定序列与uei校准物上的限制性位点是相同的,并且仅使用一种限制酶来消化实体鉴定序列和uei校准物。在分子鉴定dna序列包含产生突出端的限制性位点的实施方式中,该限制性位点与包含在实体鉴定序列和uei校准物上的限制性位点可以是相同或不同的,优选不同。
在另一个实施方式中,包含在实体鉴定序列与uei校准物上的限制性位点是不同的,并且使用两种不同的限制酶来消化实体鉴定序列和uei校准物。使用不同的限制性位点和具有相容末端的标记确保了大量产生的连接事件引起限制性位点的断裂。因此,所得嵌合分子不是混合物中存在的限制酶的底物,并且平衡被拉向期望连接产物的形成。在分子鉴定dna序列包含产生突出端的限制性位点的实施方式中,该限制性位点与包含在实体鉴定序列上或uei校准物上的限制性位点可以是相同或不同的。优选地,该限制性位点与包含在uei校准物上的限制性位点是相同的。
优选地,这些限制性位点是非回文的,以确保细胞鉴定序列、uei校准物和标记分子靶的结合的方向性。
在扩增之前,即融合两组液滴之前通过相容突出端组装uei序列和uei校准物的实施方式中,可以通过i)uei校准物和标记分子靶的相容突出端或(ii)实体鉴定序列和标记分子靶的相容突出端将包含uei序列和uei校准物的片段连接于标记分子靶。
可以在液滴融合之后,例如通过微微注射或液滴融合,或在液滴融合的同时,例如通过在t形接头处注射将限制酶、dna连接酶和任选的缓冲液添加到液滴中(参见例如图10)。可以在聚结点进行注射,其中电极的存在引发液滴融合和酶递送两者。
接着可以收集乳液并温育以使得可以消化和连接,从而结合uei条形码、标记分子靶和存在时uei校准物条形码。
在一个具体实施方式中,其中分子靶是核酸,优选rna,可以在并入uei条形码和任选的uei校准物条形码之后,向标记分子靶添加测序引物退火序列。特别地,所述方法还可以包含,在并入uei条形码和任选的uei校准物条形码之后,使用如下引物进行引物延伸反应,所述引物从3'末端至5'末端包含与分子靶的互补链即cdna杂交的区域和测序引物退火序列。
任选地,在其5'末端,引物还可以包含使得可以特异性捕获引物延伸反应产物的结合标签。优选地,结合标签是生物素或地高辛,更优选是生物素。
可以如上述进行引物延伸反应,并且可以使用任何已知方法如微微注射或液滴融合使反应混合物进入液滴中。或者,可以在液滴破坏和内容物回收之后在本体中进行引物延伸反应。
在完成标记过程之后,可以将液滴破坏并且可以回收其内容物,即液滴裂解物,例如以进一步分析/测序。
分析通过本发明方法标记的分子靶
在另一方面,本发明还涉及一种以单实体分辨率定量来自多个实体的一个或数个分子靶的方法,所述方法包含
根据上述本发明方法标记所述分子靶,捕获包含umi、uei和任选的uei校准物条形码的所述标记分子靶,
扩增包含umi和uei条形码以及任选的uei校准物条形码的序列,
对所述扩增序列进行测序。
上文对于本发明方法所述的所有实施方式也涵盖在该方面中。
捕获包含umi、uei和任选的uei校准物条形码的标记分子靶的步骤使得可从液滴裂解物中去除非靶向分子、未反应的实体鉴定序列和/或当存在时的uei校准物。该步骤可以使用能够特异性结合分子靶的任何捕获分子如特异于分子靶的抗体或核酸进行,该捕获分子与支持物连接。捕获分子可以直接或间接与支持物连接。特别地,捕获分子可以包含与支持物连接的结合标签,例如生物素,该结合标签与配偶体例如抗生蛋白链菌素相互作用。捕获分子可以是例如特异于靶向蛋白的生物素化单克隆抗体,或对特异于从靶向rna产生的cdna的合成dna寡核苷酸。可以在第二cdna链的合成期间通过上述引物延伸添加结合标签。可以选择支持物以使得可洗涤未反应的分子。优选地,支持物是珠粒,更优选抗生蛋白链菌素缀合的珠粒。在一些优选实施方式中,支持物是磁性珠粒,特别是抗生蛋白链菌素缀合的磁性珠粒。
接着使用技术人员已知的任何方法,优选使用下一代测序方法对包括(i)包含umi条形码和任选的类型鉴定符的分子鉴定dna序列和(ii)包含uei条形码的实体鉴定序列的核酸进行测序。
在一些实施方式中,其中实体鉴定序列、uei校准物和标记分子靶一起形成三部分分子,待测序的核酸包括i)包含umi条形码和任选的类型鉴定符的分子鉴定dna序列,(ii)包含uei条形码的实体鉴定序列和(iii)uei校准物条形码。
在一些其它实施方式中,其中实体鉴定序列分别与uei校准物和分子鉴定dna序列结合,待测序的核酸包括(i)包含umi条形码和任选的类型鉴定符的分子鉴定dna序列以及包含uei条形码的实体鉴定序列,和(ii)包含uei条形码和uei校准物条形码的实体鉴定序列。
在一些实施方式中,其中所述核酸不包含任何测序引物退火序列,可以在测序步骤之前添加所述序列,优选通过使用包含所述序列的引物进行dna扩增或通过连接包含所述序列的寡核苷酸进行。
优选地,当分子靶是rna时,还对该rna的一部分进行测序。
本发明的主要优点之一是可以在测序步骤之前汇集所有捕获的标记分子靶,无论分子靶是什么类型(例如rna或蛋白质)。接着可以使用技术人员已知的任何方法如生物信息学分析序列以簇集所述序列并确定分子靶的绝对定量。首先,使用uei和uei校准物条形码簇集标记源自同一实体的分子的uei组。其次,使用uei条形码根据分子的来源实体簇集所述分子。第三,根据分子的类型和身份进行簇集,任选地使用类型鉴定符,最后,使用umi条形码消除冗余序列,从而以单实体分辨率获得每个分子的绝对定量。
微流体装置
在另一方面,本发明还涉及一种适用于实施本发明方法的至少一个步骤的微流体装置。
上文对于本发明方法所述的所有实施方式也涵盖在该方面中。
如本文所用,术语“微流体装置”、“微流体芯片”或“微流体系统”是指包括至少一个微流体通道的装置、设备或系统。
微流体系统可以是或包含基于硅的芯片,并且可以使用各种技术制造,所述技术包括但不限于热压花、弹性体模制、注入模制、liga、软光刻、硅制造和相关的薄膜加工技术。用于制造微流体装置的合适材料包括但不限于环烯烃共聚物(coc)、聚碳酸酯、聚(二甲基硅氧烷)(pdms)、聚(甲基丙烯酸甲酯)(pmma)和玻璃。优选地,微流体装置通过pdms中的标准软光刻技术,随后与玻璃显微镜载玻片粘合来制备。由于一些材料如玻璃的吸附一些蛋白质并且可能抑制某些生物过程的亲水或疏水性质,所以可能需要钝化剂。合适的钝化剂是本领域已知的,并且包括但不限于硅烷、氟硅烷、聚对二甲苯、正十二烷基-β-d-麦芽糖苷(ddm)、泊洛沙姆如pluronics。
如本文所用,术语“通道”是指物品(例如衬底)上或中至少部分引导流体流动的特征。术语“微流体通道”是指如下通道,其横截面尺寸小于1mm,通常小于500μm、200μm、150μm、100μm或50μm,并且长度与最大横截面尺寸的比率至少为2:1,更通常至少为3:1、5:1、10:1或更大。应注意,术语“微流体通道”、“微通道”和“通道”在本说明书中可互换使用。通道可以具有任何横截面形状(圆形、椭圆形、三角形、不规则形、正方形或矩形等)。优选地,通道具有正方形或矩形横截面形状。可以部分或全部覆盖或敞开通道。如本文所用,术语通道的“横截面尺寸”垂直于流体流动方向测量。
本发明的微流体装置包含
-第一乳液再注入模块或芯片上液滴产生模块;
-第二乳液再注入模块或芯片上液滴产生模块
-液滴配对模块,和
-耦合液滴融合与注入的模块,
其中乳液再注入模块和/或芯片上液滴产生模块与液滴配对模块流体连通并且在液滴配对模块的上游,液滴配对模块与耦合液滴融合与注入的模块流体连通并且在耦合液滴融合与注入的模块的上游。
如本文所用,术语“上游”是指组件或模块处于在微流体系统中从给定参考点出发的流体流动的相反方向上。
如本文所用,术语“下游”是指组件或模块处于在微流体系统中从给定参考点出发的流体流动的方向上。
在一个实施方式中,本发明的微流体装置包含
两个乳液再注入模块
液滴配对模块,和
耦合液滴融合与注入的模块。
此种微流体装置的一个示例性实施方式如图10所呈现。
在另一个实施方式中,本发明的微流体装置包含
乳液再注入模块和芯片上液滴产生模块,
液滴配对模块,和
耦合液滴融合与注入的模块。
在又一个实施方式中,本发明的微流体装置包含
两个芯片上液滴产生模块,
液滴配对模块,和
耦合液滴融合与注入的模块。
技术人员可以基于任何已知技术容易地设计乳液再注入模块。通常,乳液再注入模块包含ψ形结构,其中注入的液滴由通过至少一个、优选两个与再注入通道连接的侧通道供应的载体油间隔开。
技术人员可以基于任何已知技术容易地设计用于产生液滴的模块。例如,可以通过技术人员已知的任何技术在液滴产生模块中制备乳液液滴,所述技术例如共流动流、t形接头中的交叉流动流中的小滴破裂(参见例如wo2002/068104)和流体动力流聚焦(由christopher和anna,2007,j.phys.d:appl.phys.40,r319-r336综述)。
如上文所解释,液滴配对模块是尺寸使得在两组液滴之间可发生接触的通道。在一个实施方式中,通道的宽度约为较大液滴的直径,并且通道的深度小于较大液滴的直径。在另一个实施方式中,通道的深度约为较大液滴的直径,并且通道的宽度小于较大液滴的直径。
配对通道的长度必须足以获得第一组和第二组的液滴之间的接触。优选地,接触时间大于1ms,优选大于4ms。如本文所用,“接触时间τ”是指配对的液滴在到达配对通道的末端之前保持物理接触的时间。通常,配对通道的长度在100μm至10mm范围内,优选在500μm至2mm范围内,并且更优选约为1.5mm。
耦合液滴融合与注入的模块优选是如下模块,其中配对之后的液滴由于电极的接近而暴露于使其界面不稳定的电场,并且同时与注入通道中的料流接触。接着,界面的去稳定化不仅引起液滴聚结,而且将注入的料流输入液滴中。
微流体装置还可以包含收集模块,在其中回收融合的液滴。
任选地,本发明的微流体装置可以包含在耦合液滴融合与注入的模块的下游并且在收集模块上游的入口。可以使用该入口注入额外的表面活性剂,以提高融合的液滴的稳定性和防止储存期间的任何聚结。
在另一方面,本发明还涉及一种包含根据本发明并且如上所述的微流体装置的试剂盒。
试剂盒还可以包含一个或数个微流体芯片,所述微流体芯片包含芯片上液滴产生模块。
本发明的试剂盒还可以包含
-一个或数个上述探针;和/或
-一个或数个上述实体鉴定序列;和/或
-一个或数个上述uei校准物;和/或
-一个或数个适合于扩增实体鉴定序列和/或uei校准物的引物;和/或
-水相和/或油相;和/或
-提供此种试剂盒的使用指南的活页。
上文对于本发明的方法和微流体装置所述的所有实施方式也涵盖在该方面中。
在另一方面,本发明还涉及一种试剂盒,该试剂盒包含
-一个或数个上述探针;和/或
-一个或数个上述实体鉴定序列;和/或
-一个或数个上述uei校准物;和/或
-一个或数个适合于扩增实体鉴定序列和/或uei校准物的引物;和/或
-水相和/或油相;和/或
-一个或数个微流体装置,特别是一个或数个上述微流体装置,和任选的
-提供此种试剂盒的使用指南的活页。
上文对于本发明的方法和微流体装置所述的所有实施方式也涵盖在该方面中。
本发明还涉及本发明试剂盒用于根据本发明方法标记来自多个实体的多个分子靶或用于根据本发明方法定量来自多个实体的一个或数个分子靶的用途。上文对于本发明的方法、微流体装置和试剂盒所述的所有实施方式也涵盖在该方面中。
如本文所用,动词“包含”以其非限制性含义使用,是指包括该词后面的项,但不排除未特别提及的项。
另外,除非上下文明确要求存在一个且仅一个元件,否则对未指示数量的元件的指称不排除存在超过一个所述元件的可能性。因此,未指示数量通常是指“至少一个”。
如本文所用,术语“约”是指值±指定值的10%的范围。例如,“约20”包括±20的10%,或18至22。优选地,术语“约”是指值±指定值的5%的范围。
本说明书中引用的所有专利和参考文献都在此以全文引用的方式并入。
实施例
i.微流体芯片的制造和操作
在下文提供的所有实施例中,使用相同的程序制造微流体芯片,并且使用相同的工作站对其进行操纵。
a.微流体芯片的制备和操作
使用如先前在(mazutis等人,2009)中所述的经典复制模制方法获得微流体装置。简言之,在autocad(autodesk2014)上设计装置,印刷负性光掩膜(selbas.a.)并将其用于通过标准光刻法来制备模具。使用su8-2010和su8-2025光阻剂(microchemcorp.)将10至40μm深的通道图案化到硅晶片(siltronix)上。接着使用常规的软光刻法(xia和whitesides,1998)在聚二甲基硅氧烷(pdms,silgard184,dow-corning)中制造微流体装置。等离子体结合到玻璃载玻片上之后,将通道用1%(v/v)1h,1h,2h,2h-全氟癸基三氯硅烷(97%,abcrgmbhandco.)在hfe7500(3m)中的溶液钝化,随后用压缩空气吹洗。微流体装置的关键尺寸和深度在相关图式上和其图注中给出。
将水相加载到i.d.0.75mmptfe管(thermoscientific)中,并且将油加载到2mlmicrotubes(thermoscientific)中。使用配备flowells(7μl/min流量计)的7巴mfcstm压力驱动流量控制器(fluigent)以恒定且高度控制的流速将液体注入微流体装置中,从而允许以流速控制模式操作。
b.光学装置,数据采集和控制系统
光学装置基于安装在减振平台(thorlabsb75150ae)上的倒置显微镜(nikoneclipseti-s)。使用二色镜(semrock2f495-di03-2536)组合488nm激光(crystalaserdl488-050-o)和561nm激光(coboltdpl561-nm-100mw)的光束。使用第二二色镜(semrockdi02-r405-25x36)将它们进一步与375nm激光(crystalaserdl375-020-o)组合,随后使用一对透镜(semrocklj1878l2-a和lj1567l1-a)将所得组合光束成形为线,该组合光束被引导到显微镜物镜(nikonsuperplanfluor20xelwd或nikonsuperplanfluor40xelwd)中而聚焦在检测点处的通道中间。通过同一物镜收集发射的荧光,并通过多边缘二色镜(semrockdi01-r405/488/561/635-25x36)与激光束分离。通过第三二色镜(semrocklm01-480-25)从绿色(syto9,fam)和橙色(texas-red)荧光中分解出蓝色(7-氨基香豆素-4-甲烷磺酸)荧光。接着通过另一个二色镜(semrockff562-di03-25x36)将绿色荧光与橙色荧光分离。最后通过配备有带通滤光片(对于蓝色、绿色和橙色检测,分别为semrockff01-445/45-25,ff01-600/37-25和ff03-525/50-25用于蓝色,绿色和橙色检测)的三个光电倍增管(hamamatsuh10722-20)测量荧光。使用智能数据采集(daq)模块从pmt进行信号采集,该模块具有由内部开发的固件和软件驱动的用户可编程fpga芯片(nationalinstrumentspci-7851r)。为了监测实验,使用另一个二色镜(semrockff665-di02-25x36)将光分流到ccd相机(alliedvisiontechnologiesguppyf-033)。长通滤光器(semrockblp01-664r-25)防止激光的潜在破坏性反射进入相机。
ii.实施例中使用的序列
表1:实施例中使用的序列
所有寡核苷酸(除了通过引物延伸获得的15,16和22)都购自integrateddnatechnologies(idt)。对应于uci或校准物的随机序列由n阵列表示并加下划线,而umi由斜体的n阵列表示。由alwni(cagnnnctg)和draiii(cacnnngtg)消化的序列以粗体显示。
*这些寡核苷酸被5'磷酸化以使得其可通过t4dna连接酶与另一dna连接。
**[bg]:苄基-鸟嘌呤;isp18:18个碳长的内部间隔子。
实施例1:细胞制备、个体化和裂解
在该实施例中,我们证明细菌细胞可以被个体化在液滴中并使用洗涤剂有效裂解。使用用携带在t7rna聚合酶启动子的控制下的gfp基因的质粒或用带有不相关构建体的质粒转化的大肠杆菌细菌(t7-xpresslys/iq,无gfp菌株newenglandbiolabs)作为模型细菌并且之后将它们分别归纳为菌株gfp+和gfp-。请注意,两种质粒都赋予细菌氨苄青霉素抗性,从而使其可共培养。
a.细胞的制备
首先通过接种补充有0.1mg/ml氨苄青霉素和2%葡萄糖的2yt培养基并在37℃和搅拌下温育过夜获得细胞预培养物(起始物)。接着使用500μl该起始物(od600为3.77)接种20ml相同的培养基,并且使细菌生长直至培养物达到od6000.6。接着,对细胞进行荧光染色以使得可在微流体步骤期间监测细胞。为此,根据供应商的建议,使用5μmsyto9(lifetechnologies的分子探针,参考号34854)将2ml该培养物在黑暗中染色2小时。用pbs缓冲液1×洗涤细胞1次,接着沉淀以用于进一步实验。
b.细胞包封和裂解
实验程序
将标记细胞的丸粒在补充有37.5μg/ml葡聚糖texasred70000mw(用作液滴跟踪剂)和0.05%pluronicf68的
实施例2:制备唯一鉴定符和液滴标志
在该实施例中,我们显示了如何在本体中和在微流体液滴中制备唯一鉴定符(ui),以及如何使用所述唯一鉴定符来产生编码液滴身份的标志。ui由随机序列对(uei(“唯一实体鉴定符”)和uei校准物)组成,该随机序列对由涵盖产生相容末端的限制性位点的恒定序列区包围,所述相容末端随后用于将两个序列再组合在一起(图15)。为了形成ui,应该首先在双重pcr中扩增uei校准物和uei序列,接着通过特定限制/连接反应再组合在一起。接着,包含在同一液滴中的ui库构成液滴的标志,随后可以使用该标志将所给编码分子再分配到其所来源的液滴。
a.两个条形码组的双重pcr共扩增
我们首先验证了在管中进行uei校准物和uei序列的双重pcr共扩增的可能性,接着将其以液滴微流体形式转移。
实验程序
将稀释到0.2mg/ml酵母总rna(ambion)中的500微微微摩尔(attomole)的一种或两种模板(即uei-draiii(1)和alwni-校准物-draiii(4))引入含有0.2μm每种正向引物(分子2和分子6)和反向引物(分子3和分子5)、0.2mm每种dntp、0.1%pluronicf68、0.67mg/ml葡聚糖texasred(invitrogen)、6nm实验室制备的taq聚合酶和1x
结果
扩增之后,在8%聚丙烯酰胺天然凝胶上容易观察到具有预期大小的pcr产物(对于uei-draiii为62个碱基对,并且对于alwni-校准物-draiii为142个碱基对)。然而,当仅存在一种模板时观察到单个条带(图16,泳道1和2),当将两种模板混合在一起时看到两个不同的条带(图16,泳道3),从而证实两种模板都可以有效地共扩增。在该部分中,只有校准物带有随机化区域,这解释了与校准物相对应的带的略微拖尾。这个拖尾归因于在两个具有相同恒定区但具有不同随机化区的不同校准物的链之间形成异源双链体。
b.油包水液滴中pcr的扩增
我们接下来验证共扩增可以在液滴中有效地发生。
实验程序
将稀释到0.2mg/ml酵母总rna(ambion)中的14微微微摩尔(使得理论平均值可为每个液滴4个模板dna分子的浓度)每种模板(即模板uei-n15-draiii(7)和模板alwni-n15-校准物-draiii(8))引入含有0.2μm每种正向引物(分子2和分子6)和反向引物(分子3和分子5)、0.2mm每种dntp、0.1%pluronicf68、0.67mg/ml葡聚糖texasred(invitrogen)、6nm实验室制备的taq聚合酶和1x
结果
凝胶分析证实双重pcr扩增在油包水液滴中正确地进行(图16,泳道5)并且具有与在本体中相同的表观效率(图16,泳道4)。此外,分析显示将反应区室化在液滴中使得可获得分辨更好的条带,可能是通过限制重组、异源双链体的形成和与两个条形码(uei校准物和uei)上随机化区域的存在相关的其它pcr人工产物。
c.基于ui的标志
形成ui需要通过限制/连接耦合反应将uei校准物和uei重新组合在一起。为此,我们选择两个非回文限制性位点,从而产生相容的3'突出端。alwni(5'-cagnnn/ctg-3')和draiii(5'-cacnnn/gtg-3')是满足这些标准的两种酶消化序列(在表1中加粗)。在这些位点的消化产生两个相容的序列,所述序列可以重新组合在一起并且形成新的序列(即5'-cagnnngtg-3'和5'-cacnnnctg-3'),这些新的序列不再能被所述酶消化,因此将平衡拉向重组分子的形成。
实验程序
使用模板分子7和8以及扩增引物3、5和6重复在部分b中建立的双重pcr。但将引物2换成引物9,这使得可引入illu-1序列,该序列是随后用于下一代测序分析的衔接子序列。为了评价两种条形码的相对量可以影响重组效率,从而影响ui形成的程度,我们使用在液滴中两种相对浓度的uei校准物和uei进行了一组实验。通过改变pcr混合物中相应引物的浓度来改变该比率。但通过以每个pcr液滴相同的平均模板数开始每个实验而保持条形码的多样性。因此,在第一反应组中,将稀释到0.2mg/ml酵母总rna(ambion)中的14微微微摩尔每种模板与0.2μm每种引物(3、5、6和10)混合,得到1/1的最终uei校准物/uei比率。在第二反应组中,将稀释到0.2mg/ml酵母总rna(ambion)中的14微微微摩尔每种模板与0.2μm引物3和引物10以及0.02μm引物5和引物6混合,得到0.1/1的最终uei校准物/uei比率。接着将每种模板/引物混合物引入200μl含有0.2mm每种dntp、0.1%pluronicf68、0.67mg/ml葡聚糖texasred(invitrogen)、6nm实验室制备的taq聚合酶和1x
最后,使用基于python的生物信息学算法分析序列(图20)。该算法分三个主要步骤。首先,将从测序仪获得的原始序列进行质量过滤,并且将q值低于30的原始序列从库中去除。此外,还从库中去除了在非随机化区域中呈现突变或显示不适当长度的序列。其次,提取uei校准物和uei序列,并且将来自同一液滴的对簇集在一起。简言之,将与给定uei校准物相关的所有uei都簇集在一起。接着,将共享包含在同一群集中的uei的所有uei校准物也簇集在一起。在该过程结束时,获得ui群集(uei校准物和uei对)并且形成液滴的标志。最后,在第三步骤中,可以使用所述标志将来自库的每个分子再分配到其所来源的液滴。
结果
ui标志仅在限制/连接酶存在下容易形成。实际上,qpcr分析显示无论使用哪种uei校准物/uei比率,在重组酶不存在的情况下都需要超过10个额外的扩增循环(δct>10)来达到阈值(图19,上图,比较-和+酶栏),从而指示在酶不存在的情况下,重组物质至少减少了一千倍。此外,电泳凝胶分析(图19,左图)显示预期大小的特定条带仅在酶存在下获得(泳道2、4和7)。因此,ui形成受到高度控制,因为它仅在特定酶的存在下发生并且在pcr反应期间不形成自发形式,这限制了以非特异性方式形成ui的风险。发现这种重组不仅可在管中进行(图19,左图,泳道1至4),而且其也可在液滴中进行(图19,左图,泳道5至7),从也在液滴中制备的pcr扩增产物开始。与在管中一样,在液滴中存在ui需要存在特定的限制/连接酶(比较泳道6和7)。
通过下一代测序以及图20所示的生物信息学算法进一步分析含有ui的液滴,使得我们可将每个uei和每个uei校准物再分配到其所来源的液滴(图21)。实际上,使用由15个随机化(109个不同变体的理论多样性)区域组成的条形码使得不可能在乳液(由最多几百万个液滴组成)的超过一个液滴中找到相同的uei或相同的uei校准物,从而使这种再分配可靠。每个液滴的不同uei和uei校准物的数量遵循泊松分布(poissondistribution)(图21,左;还参见图23),如对于随机分布到区室如液滴中的对象所预期。这证实了在整个过程中保持了信息的适当限制,并且ui是在液滴中而不是在液滴破坏之后产生的。请注意,在该实施例中,每个液滴的不同uei校准物的平均数量仅为1,而预期为4。然而,此后呈现的补充实验(部分d)使得可再调整该参数并进一步证实分子的泊松驱动行为。
簇集每个液滴中含有的不同ui使得可以获得对于每个液滴唯一的标志。有趣的是,在序列库中均匀地呈现标志(图21,右),指示在ui制备期间没有显著的液滴与液滴偏差。
d.调整uei多样性
为了进一步证实uei校准物和uei两者遵循泊松分布在液滴中分布,以及适当地调整每种类型条形码的含量,以每个液滴三种不同平均数量(λ值)的条形码进行实验。
实验程序
如前所述进行双重pcr,但使用不同起始量的模板分子7和8。为了测试λ1.3条件(即每个液滴平均有1.3个uei校准物和1.3个uei),将稀释到0.2mg/ml酵母总rna(ambion)中的5微微微摩尔的每种模板(即模板uei-n15-draiii(7)和模板alwni-n15-校准物-draiii(8))引入含有0.2μm每种正向引物(分子2和分子6)和反向引物(分子3和分子5)、0.2mm每种dntp、0.1%pluronicf68、0.67mg/ml葡聚糖texasred(invitrogen)、6nm实验室制备的taq聚合酶和1x
结果
在改变每个液滴的uei校准物和uei模板的初始数量(λ值)的情况下在液滴中制备ui并不会显著挑战过程本身。实际上,无论起始λ值是多少(1.3至12),当定量在该过程(图22中的反应2至3)中扩增并重新组合到液滴中的dna的量时,都获得了非常接近的ct值。相对于无酶对照(图22中的反应1),在添加酶时达到阈值需要更少多达20个扩增循环,从而指示在酶存在下形成高达100万倍的ui。对凝胶的分析证实,仅在酶存在下观察到正确的反应产物(图22中的箭头所示)(泳道2至4),而在酶不存在的情况下仅获得少量寄生性扩增产物(泳道1)。
使用我们的生物信息学算法分析来自每种条件的1000个液滴的等分试样的序列含量揭示,如所预期,改变每个液滴的uei校准物和uei模板的起始量直接影响液滴中两种条形码的分布(图23),并且每个液滴的不同uei校准物和uei的起始数越高,形成液滴标志的不同ui的数量越多。因此,调整两种模板的初始浓度允许以可预测方式调整每个液滴的ui的多样性(因此标志的复杂性)。
实施例3:使用ui/标志来标记和定量dna
因为ui是通过消化/连接过程获得的,因此它们也可以附加到另一个dna分子上,条件是该分子具有与限制产物之一相容的位点。该dna可以是逆转录产物或与另一个分子如蛋白质连接的dna。在该实施例中,我们证明了可以将ui与另一个dna连接,并且随后使用我们的方法将每个dna分子再分配到其所来源的液滴,以及定量每个液滴的dna含量。因此,在该实施例中,我们旨在:i)分离和pcr共扩增uei校准物和uei模板;ii)用呈现与限制产物之一相容的末端的dna补充每个含有uei校准物/uei的液滴;iii)向每个液滴添加限制/连接酶以通过重组形成ui并将其连接在靶dna分子的末端;iv)以高通量方式对文库进行索引化和测序。此外,将形成唯一分子鉴定符(umi)的8个核苷酸的随机序列插入靶dna中以使得可随后计数每个液滴中含有的分子。
a.在管中进行uei形成和dna标记
在液滴中进行dna标记之前,我们首先在管中验证了整个反应过程。在该实施例中,我们使用模拟逆转录产物的部分双链dna(图24)。该分子由长分子(13)组成,其3'部分保持单链并在其5'部分带有8个核苷酸的随机化区域(umi),之后是通过与分子14配对形成双链的恒定区。该配对产生与ui的uei校准物部分的draiii切割产物相容的3'突出端。此外,13的5'磷酸化末端使其成为dna连接酶的底物。
实验程序
首先如实施例2中通过双重pcr共扩增来产生ui组分(uei校准物和uei)。为此,如实施例2部分a中所述,首先使用0.2μm引物2、3、5和12,以200μl的体积pcr扩增500微微微摩尔模板dna7和11。通过将1.25pmol分子13和1.25pmol分子14在20μl
结果
分子13和14的退火形成靶dna,其呈现与在uei校准物消化之后产生的draiii产物相容的3'突出端。将该靶dna与共扩增的uei校准物和uei以及限制酶和连接酶混合在一起使得可形成ui并使其与靶dna连接,从而直接对其进行编码。qpcr分析指示在重组酶不存在的情况下需要超过17个额外的扩增循环(δct>17)来达到阈值(图25,比较-和+酶栏),从而指示在酶不存在的情况下,ui编码的dna分子至少减少了160,000倍。此外,凝胶电泳分析(图25,右图)显示预期大小的特定条带仅在酶存在下获得(泳道2)。
因此,可以使用ui来编码显示与ui中存在的限制性位点之一相容的突出端的dna。
b.在液滴中进行ui形成和dna标记
接着我们将dna标记转换成液滴微流体形式。如在实施例2中,使用双重pcr在液滴中共扩增uei校准物和uei,接着添加dna以进行标记并且向每个液滴添加酶混合物。
实验程序
首先通过引物延伸制备分子7的包含测序衔接子illu1的延伸形式。简言之,将10pmol模板7与50pmol引物9在50μl含有0.2mm每种dntp、1uphusiondna聚合酶(thermoscientific)和建议工作浓度的相应缓冲液的反应混合物中混合。接着将混合物在98℃下温育5分钟并在72℃下温育10分钟。接着在琼脂糖凝胶上纯化延伸产物,并且用wizardsvgel和pcr净化系统(promega)回收dna。延伸分子现在称为模板uei-n15-draiii-illu1并且对应于分子15。将稀释到0.2mg/ml酵母总rna(ambion)中的14微微微摩尔(使得平均值为每个液滴4个模板dna分子的浓度)的每种模板(即模板uei-n15-draiii-illu1(15)和模板-alwni-n15-calib-draiii(11))引入含有0.2μm每种正向引物(分子2和分子5)和反向引物(分子10和分子12)、0.2mm每种dntp、0.1%pluronicf68、0.67mg/ml葡聚糖texasred(invitrogen)、6nm实验室制备的taq聚合酶和1x
结果
在该实施例中,我们制备了由20pl液滴组成的乳液,每个液滴平均含有约1000个靶dna分子。同时,如实施例2中所述制备含有uei校准物/uei的液滴。最后,使用我们的条形码添加微流体装置(图27),将每个含有靶dna的液滴与单个含有uei校准物/uei的液体融合,并接收控制量的限制/连接酶混合物。通过对约1000个液滴的内容物进行测序分析,我们应用了我们的生物信息学算法的升级版(图29)并且成功地:i)鉴定了样品中含有的不同ui并将其簇集成液滴标志,ii)使用这些标志将每个序列再分配到其所来源的液滴,最后iii)计数每个液滴中存在的靶dna拷贝数(图30)。该分析使我们鉴定出约1200个不同的液滴,该值非常接近1000个预期的液滴。此外,在每个液滴中平均发现靶dna的约200个拷贝,该值再次非常接近理论液滴含量(预期每个液滴约1000个拷贝)。dna拷贝数分布非常紧密,从而指示不同液滴之间条形码添加过程(即ui的形成和添加)的高度均一性。在序列池中发现每个序列-umi-ui至少10次,从而指示以10倍覆盖率分析该文库。这指示液滴含量的紧密分布确实是由所述方法的饱和引起的,并且相当代表了乳液的实际含量。总之,该实施例中呈现的结果显示,我们的方法和分析算法使得可通过将ui与液滴的dna连接来以可再现方式定量液滴的dna含量。
实施例4:将rna转化为可由ui编码的cdna
由于我们的方法使得可标记和计数乳液液滴中含有的dna分子,所以我们接下来研究了rna是否可以在液滴中转化为可用作我们的标记技术的底物的cdna。为了产生此种cdna,我们设计了由以下组成的复合逆转录(rt)引物:i)双链5'部分,其由与ui末端存在的限制性位点相容的3'突出端封端;接着ii)含有8个随机核苷酸并且用作唯一分子标识符(umi)的单链区域;和iii)用于封端的3'单链区域,其与靶rna特异性退火(图31)。在该实施例中,我们显示可以使用此种分子引发rna的逆转录以及ui的连接点。
a.在液滴中进行rna的逆转录
为了测试在液滴中逆转录rna的可能性,我们选择了来自金黄色葡萄球菌(staphylococcusaureus)的高度结构化(因此难以逆转录)rnaiii(benito等人,2000)。通过使用(ryckelynck等人,2015)中所述的相同程序使用t7rna聚合酶进行体外转录来制备rna。转录之后,在尺寸排阻柱nap5(ge-healthcare)上纯化rna,并使用nanodrop装置定量。为了防止rna在包封在液滴中之前的不想要的逆转录,首先将rna和rt试剂分别乳化并通过液滴融合混合在一起(mazutis等人,2009)。此外,为了评价逆转录过程的灵敏性,我们制备了每个液滴含有10、100或1000个rna分子的乳液。接着向每个液滴中添加逆转录物,并使rt可继续进行,随后分析所产生的cdna。
实验程序
将0.6、6或60毫微微摩尔rna-iii(使得每2pl液滴分别具有10、100或1000个rna分子)引入100μl含有
结果
在该实施例中,制备2pl各自含有10、100或1000个rna-iii分子的液滴的乳液,并与18pl含有rt混合物的液滴融合。作为对照,以本体形式进行相同实验。在温育之后,qpcr分析显示在本体和乳化形式下,rt均以相同的效率进行(图34)。实际上,在两种形式下,通过本体和乳化反应获得的ct值非常接近并且显著高于没有逆转录酶的对照反应的ct值。此外,凝胶分析证实在两种条件下获得的qpcr产物具有预期的大小,而阴性对照仅获得小尺寸的pcr副产物。有趣的是,在rna稀释最多的情况下(每2pl10个分子)仅在乳液中给出了对应于预期大小的扩增产物的可检测信号,从而证明液滴形式提供更高灵敏性并且表明如果以至少10个存在靶rna,则应该可以将其转化为呈现与ui的接枝相容的末端的cdna。实际上,在该实施例中,我们验证了使用具有序列元件的rt引物使得可使用实施例3中验证的策略有效地接枝ui(比较图24和31)。
实施例5:探索uei校准物和uei序列的替代形式
我们注意到,当在低浓度的靶分子下工作时,在一些引物与uei校准物和uei的随机化区域之间发生了一些非特异性重组事件。因此,我们探索了15个连续随机化核苷酸的简单片段的替代序列。在该实施例中,我们逆转录纯化的gfpmrna,接着用从如下模板产生的ui对所获得的cdna进行标记:i)不含随机化区域;ii)或带有15个连续随机化核苷酸的片段(n15);iii)或带有半随机化核苷酸的片段,其中5个随机化二核苷酸由恒定的二核苷酸间隔开(n2x5);iv)或带有半随机化核苷酸的片段,其中4个随机化三核苷酸由恒定的三核苷酸间隔(n4443)。
gfprna的逆转录和ui添加
实验程序
通过使用(ryckelynck等人,2015)中所述的相同程序使用t7rna聚合酶进行体外转录来制备gfpmrna。转录之后,在尺寸排阻柱nap5(ge-healthcare)上纯化rna,并使用nanodrop装置定量。将16.67毫微微摩尔纯化的gfpmrna在20μl补充有1.25pmolrt引物(分子23)、1.25pmol互补寡核苷酸(分子24)、1mmdntp、10mmdtt、10uamv逆转录酶、0.1μmfam的以建议浓度制备的
结果和结论
正确地逆转录靶rna(gfpmrna),并将4种类型的条形码(uei校准物/uei)附加到所得cdna上。尽管去除了随机化区域或提供n2x5半随机化区域的条形码产生了均匀的pcr产物(图35中的泳道1和2),但在n4443和n15区域的情况下观察到更多的拖尾条带。此外,在n4443和n15的情况下倾向于显著累积较小尺寸的二次扩增产物。因此,半随机化条形码如n2x5的代表了更常规的n15条形码设计的有吸引力的替代方案。
实施例6:用ui标记蛋白质
为了将该技术的使用扩展到核酸以外的分子,我们在该实施例中验证了我们的方法编码与蛋白质共价连接的dna片段的能力。为此,我们克隆了由蛋白a和snap标签(newenglandbiolabs)编码区组成的融合基因。使所得基因在大肠杆菌中过表达,并且纯化所得蛋白质(nabab)。在该实施例中,我们首先验证了唯一的dna片段可以与蛋白质共价连接,并且其不会干扰蛋白质通过其蛋白a部分与抗体相互作用的能力。接着,我们测试了ui是否可以与蛋白质所附加的dna链连接。
a.以每个抗体单个dna标记抗体
我们制备了一种构建体,其中编码蛋白a的序列与his标记形式的snap-标签(newenglandbiolabs)融合,并将该构建体置于表达质粒上。接着在大肠杆菌中过量产生相应的蛋白质,并通过亲和色谱法纯化。snap结构域是与呈现苄基-鸟嘌呤的底物分子反应的催化模块。反应之后,单个底物分子与snap模块共价且不可逆地连接(图36)。此外,这种用单个dna分子修饰的蛋白质也应能够通过其蛋白a结构域与抗体强烈结合。在这项研究中,我们显示我们的融合蛋白确实能够用单个dna分子标记,并且所得复合物仍然能够与抗体相互作用。
实验程序
使用用苄基-鸟嘌呤(bg)基团修饰以使得可将dna接枝在nabab的snap模块上的引物(分子32)和用alexa-488荧光基团修饰以荧光标记dna的引物(分子33)pcr扩增具有唯一alwni以及8个随机化位置的umi的模板dna(分子31)。将10pmol模板31与1nmol每种引物(32和33)在1ml含有0.2mm每种dntp、20uq5dna聚合酶(newenglandbiolabs)和建议浓度的相应缓冲液的pcr溶液中混合。接着将混合物放置在热循环仪中,并且进行在98℃下30sec的初始变性步骤,接着25个在98℃下10sec、在55℃下10sec和在72℃下30sec的循环。最后,通过在72℃下2min的最终延伸步骤结束程序。使用试剂盒wizardsvgel和pcr净化系统(promega)纯化扩增产物并且用nanodrop装置定量。接着将200pmolbg/alexa488双重标记的dna与180pmol纯化的nabab在1ml以建议浓度(1×)稀释并且补充有1mmdtt的
在第二个实验中,将27pmol纯化的nabab与20pmolbg/alexa488双重标记的dna在40μl以建议浓度(1×)稀释并且补充有1mmdtt的
结果和结论
将bg/alexa488双重标记的dna与略过量的nabab一起温育,从而使得可将所有dna接枝到nabab的snap模块上,这通过凝胶上dna条带的完全上移证明(图37,左图)。在igg存在下形成nabab-dna共价复合物显示可以容易地形成nabab-dna/igg三元复合物(图37,右图,泳道3至5)。此外,考虑到igg的平均分子量为约150kda,可以看出27pmolnabab-dna复合物在等摩尔量的igg(125mg/ml或30pmoligg,泳道4)存在下完全转移,而使用一半igg的情况下只有一半的复合物转移。这些数据指示,nabab不仅可以与单个dna分子共价连接,而且将其按化学计量与igg混合可形成nabab-dna/igg三元复合物,其中一个抗体分子主要用单个dna分子标记。
b.用uei标记nabab-dna复合物
我们研究了使用实施例3中呈现的标记策略用ui标记nabab-dna复合物的可能性。
实验程序
如实施例3-部分a中所述,在200μl补充有0.2μm每种引物(2、5、10和12)的pcr混合物中共扩增500微微微摩尔模板uei校准物和uei(分子11和15)。在热循环之后,将10μl双重pcr产物添加到10μl以建议浓度(1×)稀释且补充有2pmolnabab-dna复合物(如该实施例的部分a中所述制备)、2mmratp、20mmdtt、2.5draiiihf(newenglandbiolabs)、2.5u/μlalwn1(newenglandbiolabs)、25u/μlt4dna连接酶(newenglandbiolabs)、10μm乙酸香豆素和0.1%pluronicf68的
结果和结论
在该部分中,我们显示使用我们的限制/连接策略与nabab蛋白共价结合的dna可以用作ui受体。实际上,在酶不存在的情况下,需要超过26个额外的扩增循环(δct>26)来达到阈值(图38,比较-和+酶栏),从而指示在酶不存在的情况下,形成的ui-靶dna至少减少了3300万倍。此外,凝胶电泳分析(图38)显示预期大小的特定条带仅在酶存在下获得(泳道2)。
总之,在该实施例中,我们证明了可以使用nabab融合蛋白来用唯一dna分子特异性标记igg,并且即使该dna与nabab蛋白共价连接,还可以使用我们的限制/连接策略将该dna用作ui受体。此外,因为在天然条件下发生重组以保持非共价复合物(例如抗体/抗原)的结合,所以nabab-dna/igg复合物和ui编码的组合使用可以用于标记和编码igg识别的靶标(例如蛋白质)并通过下一代测序(例如miseq)对所述靶标进行定量。
总结和结论
本文报道的不同实施例证明了本发明方法的完全技术可行性。实际上,我们证明了在本体(实施例2.a)和乳化反应(实施例2.b)中,可以在双重pcr中有效地共扩增唯一鉴定符(ui)的两个组分(uei校准物和uei),接着通过限制/连接反应重新组合在一起以形成ui。接着可以将液滴中含有的ui库用作标志,从而使得可明确地将下一代测序数据库中含有的序列分配到其所来源的液滴(实施例2.c)。此外,显示了改变不同模板uei校准物和uei的初始值使得可调整标志的复杂性(实施例2.d)。
此外,我们证明了在本体中(实施例3.a)和在液滴中(实施例3.b),这些ui还可以与呈现与ui上存在的限制性位点相容的3'突出端的靶dna分子连接。同样,使用基于ui的标志使得可以以大的液滴与液滴可再现性明确地将每个dna序列分配到其所来源的液滴。
此外,我们显示可以在液滴中使用逆转录引物将rna有效地逆转录成cdna(实施例4.a),所述逆转录引物能够在之后与ui耦合(实施例4.b)或在用于逆转录靶rna之前与ui缀合(实施例5)。在这两种情况下,获得了类似的rt和ui耦合产率。我们的编码策略不限于核酸,因为我们显示我们的ui接枝策略还可以应用于用具有限制性位点的单个dna分子标记的igg,所述限制酶位点使得该dna分子与我们的限制/连接方法相容(实施例6)。最后一种情形使得可以以保持非共价生物复合物(例如抗体/抗原复合物)的方式编码非核酸靶标如蛋白质。
最后,我们显示可以分离细胞并裂解成小的4pl液滴(实施例1)。不仅物质可以从细胞中释放,而且液滴的大小使得可与液滴融合装置的使用直接相容(图33)。因此,可以直接向细胞提取物补充试剂,从而使得可逆转录靶rna和/或与单个dna分子耦合的抗体。在逆转录/抗原捕获之后,然后可以使用在实施例3中验证的条形码添加芯片(图27)将不同的uei校准物/uei组与限制/连接酶一起递送至每个液滴。最后,在索引化和下一代测序之后,使用我们的生物信息学算法(图29)使得可将每个序列(最初对应于rna、蛋白质或其他分子)再分配到其所来源的液滴,并且这种方式使得可定量每个细胞的含量。
参考文献
benito,y.等人,rna,2000.6(5):第668-79页.
brown,r.b.和audet,j.r.soc.interface5,s131-s138(2008)
islam等人,nat.protoc.2012;7:813-828.
kintses等人,chem.biol.2012.19,1001-1009
klein等人,cell.2015.161,1187-1201
macosko等人,cell.2015,161,1202-1214
mazutis,l.等人,analyticalchemistry,2009.81(12):第4813-21页.
novak等人,angew.chem.int.ed.50,390-395(2011)
rau等人,appl.phys.lett.2004.84,2940-2942
rotem等人,plosone,2015,10(5):e0116328
ryckelynck,m.等人,rna,2015.21(3):第458-69页.
siegel,a.c.等人,advancedmaterials,2007.19(5):第727-733页.
streets等人,proc.natl.acad.sci.usa,2014,111,7048-7053
xia,y.n.和g.m.whitesides,softlithography.angewandtechemie-internationaledition,1998.37(5):第551-575页.
序列表
<110>斯特拉斯堡大学(universitedestrasbourg)
法国国家科学研究中心(centrenationaldelarecherchescientifique)
<120>靶分子的串联条形码添加以便以单实体分辨率对靶分子进行绝对定量
<130>b2367pc
<160>37
<170>patentinversion3.5
<210>1
<211>62
<212>dna
<213>人工序列
<220>
<223>模板uei-draiii
<400>1
gagcggataacaatttcacacaggcacggggtgtgagatcacagatcggaagagcgtcgt60
gt62
<210>2
<211>24
<212>dna
<213>人工序列
<220>
<223>uei-fwd
<400>2
gagcggataacaatttcacacagg24
<210>3
<211>19
<212>dna
<213>人工序列
<220>
<223>uei-rev
<400>3
acacgacgctcttccgatc19
<210>4
<211>142
<212>dna
<213>人工序列
<220>
<223>模板校准物-n15-alwni
<220>
<221>misc_feature
<222>(86)..(110)
<223>n为a、c、g或t
<400>4
tcgtcggcagcgtcagatgtgtataagagacagggaagacgtagcaagggagtagccaat60
gtgaattgagagccttaagctgtatnnnnnnnnnnnnnnnnnnnnnnnnncaggggctgg120
tcgtgactgggaaaaccctggc142
<210>5
<211>23
<212>dna
<213>人工序列
<220>
<223>calib-rev
<400>5
gccagggttttcccagtcacgac23
<210>6
<211>19
<212>dna
<213>人工序列
<220>
<223>illu-2-fwd
<400>6
tcgtcggcagcgtcagatg19
<210>7
<211>67
<212>dna
<213>人工序列
<220>
<223>模板uei-n15-draiii*
<220>
<221>misc_feature
<222>(34)..(48)
<223>n为a、c、g或t
<400>7
gagcggataacaatttcacacaggcacggggtgnnnnnnnnnnnnnnngatcggaagagc60
gtcgtgt67
<210>8
<211>149
<212>dna
<213>人工序列
<220>
<223>模板校准物-n15-alwni-lg
<220>
<221>misc_feature
<222>(103)..(117)
<223>n为a、c、g或t
<400>8
tcgtcggcagcgtcagatgtgtataagagacagggaagacgtagcaagggagtagccaat60
gtgaattgagagccttaagctgtatcaggaccagagagatgannnnnnnnnnnnnnncag120
gggctggtcgtgactgggaaaaccctggc149
<210>9
<211>53
<212>dna
<213>人工序列
<220>
<223>illu1revuei
<400>9
gtctcgtgggctcggagatgtgtataagagacagacacgacgctcttccgatc53
<210>10
<211>19
<212>dna
<213>人工序列
<220>
<223>illu1
<400>10
gtctcgtgggctcggagat19
<210>11
<211>90
<212>dna
<213>人工序列
<220>
<223>模板-alwni-n15-calib-draiii
<220>
<221>misc_feature
<222>(44)..(58)
<223>n为a、c、g或t
<400>11
ggaagacgtagcaagggagtagccaatgtgaattcacacagtgnnnnnnnnnnnnnnnca60
ggggctggtcgtgactgggaaaaccctggc90
<210>12
<211>24
<212>dna
<213>人工序列
<220>
<223>calib-fwd
<400>12
ggaagacgtagcaagggagtagcc24
<210>13
<211>80
<212>dna
<213>人工序列
<220>
<223>rt模拟物
<220>
<221>misc_feature
<222>(36)..(43)
<223>n为a、c、g或t
<400>13
ctgcgcggatcccggaagcgaggccagctggctgcnnnnnnnnttggctgtctcttatac60
acatctgacgctgccgacga80
<210>14
<211>38
<212>dna
<213>人工序列
<220>
<223>抗sbaca-alwn1
<400>14
gcagccagctggcctcgcttccgggatccgcgcagaca38
<210>15
<211>101
<212>dna
<213>人工序列
<220>
<223>模板uei-n15-draiii-illu1
<220>
<221>misc_feature
<222>(34)..(48)
<223>n为a、c、g或t
<400>15
gagcggataacaatttcacacaggcacggggtgnnnnnnnnnnnnnnngatcggaagagc60
gtcgtgtctgtctcttatacacatctccgagcccacgagac101
<210>16
<211>539
<212>rna
<213>金黄色葡萄球菌(staphylococcusaureus)
<400>16
auuaauacgacucacuauaccuagaucacagagaugugauggaaaauaguugaugaguug60
uuuaauuuuaagaauuuuuaucuuaauuaaggaaggagugauuucaauggcacaagauau120
cauuucaacaaucggugacuuaguaaaauggauuaucgacacagugaacaaauucacuaa180
aaaauaagaugaauaauuaauuacuuucauuguaaauuuguuaucuacguauaguacuaa240
aaguaugaguuauuaagccaucccaacuuaauaaccauguaaaauuagcaagugaguaac300
auuugcuaguagaguuaguuuccuuggacucagugcuauguauuuuucuuaauuaucauu360
acagauaauuauuucuagcauguaagcuaucguaaacaacaucgauuuaucauuauuuga420
uaaauaaaauuuuuuucauaauuaauaacauccccaaaaauagauugaaaaaauaacugu480
aaaacauucccuuaauaauaaguauggucgugagccccucccaagcucgcggccuuuug539
<210>17
<211>10
<212>dna
<213>人工序列
<220>
<223>空白
<220>
<221>misc_feature
<222>(1)..(10)
<223>n为a、c、g或t
<400>17
nnnnnnnnnn10
<210>18
<211>55
<212>dna
<213>人工序列
<220>
<223>rt-umi-rnaiii*
<220>
<221>misc_feature
<222>(36)..(41)
<223>n为a、c、g或t
<400>18
ctgcgcggatcccggaagcgaggccagctggctgcnnnnnnttgggaggggctca55
<210>19
<211>10
<212>dna
<213>人工序列
<220>
<223>空白
<220>
<221>misc_feature
<222>(1)..(10)
<223>n为a、c、g或t
<400>19
nnnnnnnnnn10
<210>20
<211>35
<212>dna
<213>人工序列
<220>
<223>单独的sb
<400>20
ctgcgcggatcccggaagcgaggccagctggctgc35
<210>21
<211>63
<212>dna
<213>人工序列
<220>
<223>rnaiii-fwd
<400>21
tcgtcggcagcgtcagatgtgtataagagacagataactgtaaaacattcccttaataat60
aag63
<210>22
<211>867
<212>rna
<213>人工序列
<220>
<223>gfpmrna
<400>22
augaccagcuacccauacgauguuccagauuacgcuaucgaaggccgcggccgccaucau60
caucaucaucaugauaucgguaccaguaaaggagaagaacuuuucacuggaguuguccca120
auucuuguugaauuagauggugauguuaaugggcacaaauuuucugucaguggagagggu180
gaaggugaugcaacauacggaaaacuuacccuuaaauuuauuugcacuacuggaaaacua240
ccuguuccauggccaacacuugucacuacuuucgcguauggucuucaaugcuuugcgaga300
uacccagaucauaugaaacagcaugacuuuuucaagagugccaugcccgaagguuaugua360
caggaaagaacuauauuuuucaaagaugacgggaacuacaagacacgugcugaagucaag420
uuugaaggugauacccuuguuaauagaaucgaguuaaaagguauugauuuuaaagaagau480
ggaaacauucuuggacacaaauuggaauacaacuauaacucacacaauguauacaucaug540
gcagacaaacaaaagaauggaaucaaaguuaacuucaaaauuagacacaacauugaagau600
ggaagcguucaacuagcagaccauuaucaacaaaauacuccaauuggcgauggcccuguc660
cuuuuaccagacaaccauuaccuguccacacaaucugcccuuucgaaagaucccaacgaa720
aagagagaccacaugguccuucuugaguuuguaacagcugcugggauuacacauggcaug780
gaugaacuauacaaagagaauucagagcucggauccacucgagaugcauuagaacaaaaa840
uuauuaucagaagaagauuuaaauuaa867
<210>23
<211>69
<212>dna
<213>人工序列
<220>
<223>gfp-mut2-rt-umi-alwni*
<220>
<221>misc_feature
<222>(36)..(43)
<223>n为a、c、g或t
<400>23
ctgcgcggatcccggaagcgaggccagctggctgcnnnnnnnncaaataaatttaagggt60
aagttttcc69
<210>24
<211>26
<212>dna
<213>人工序列
<220>
<223>gfp-fwd-25nt
<400>24
tgaattagatggtgatgttaatgggc26
<210>25
<211>70
<212>dna
<213>人工序列
<220>
<223>模板-uei-draiii-n2x5
<220>
<221>misc_feature
<222>(34)..(35)
<223>n为a、c、g或t
<220>
<221>misc_feature
<222>(38)..(39)
<223>n为a、c、g或t
<220>
<221>misc_feature
<222>(42)..(43)
<223>n为a、c、g或t
<220>
<221>misc_feature
<222>(46)..(47)
<223>n为a、c、g或t
<220>
<221>misc_feature
<222>(50)..(51)
<223>n为a、c、g或t
<400>25
gagcggataacaatttcacacaggcacggggtgnnacnngannctnngcnngatcggaag60
agcgtcgtgt70
<210>26
<211>76
<212>dna
<213>人工序列
<220>
<223>模板-uei-draiii-n4443
<220>
<221>misc_feature
<222>(34)..(37)
<223>n为a、c、g或t
<220>
<221>misc_feature
<222>(41)..(44)
<223>n为a、c、g或t
<220>
<221>misc_feature
<222>(48)..(51)
<223>n为a、c、g或t
<220>
<221>misc_feature
<222>(55)..(57)
<223>n为a、c、g或t
<400>26
gagcggataacaatttcacacaggcacggggtgnnnnacannnngagnnnntctnnngat60
cggaagagcgtcgtgt76
<210>27
<211>85
<212>dna
<213>人工序列
<220>
<223>模板-校准物-alwni
<400>27
ggaagacgtagcaagggagtagccaatgtgaattcacacagtgtctacaagtacaggggc60
tggtcgtgactgggaaaaccctggc85
<210>28
<211>93
<212>dna
<213>人工序列
<220>
<223>模板-校准物-alwni-n2x5
<220>
<221>misc_feature
<222>(44)..(45)
<223>n为a、c、g或t
<220>
<221>misc_feature
<222>(48)..(49)
<223>n为a、c、g或t
<220>
<221>misc_feature
<222>(52)..(53)
<223>n为a、c、g或t
<220>
<221>misc_feature
<222>(56)..(57)
<223>n为a、c、g或t
<220>
<221>misc_feature
<222>(60)..(61)
<223>n为a、c、g或t
<400>28
ggaagacgtagcaagggagtagccaatgtgaattcacacagtgnntcnntanncannagn60
ncaggggctggtcgtgactgggaaaaccctggc93
<210>29
<211>99
<212>dna
<213>人工序列
<220>
<223>模板-校准物-alwni-n4443
<220>
<221>misc_feature
<222>(44)..(47)
<223>n为a、c、g或t
<220>
<221>misc_feature
<222>(51)..(54)
<223>n为a、c、g或t
<220>
<221>misc_feature
<222>(58)..(61)
<223>n为a、c、g或t
<220>
<221>misc_feature
<222>(65)..(67)
<223>n为a、c、g或t
<400>29
ggaagacgtagcaagggagtagccaatgtgaattcacacagtgnnnntctnnnnacannn60
ngagnnncaggggctggtcgtgactgggaaaaccctggc99
<210>30
<211>90
<212>dna
<213>人工序列
<220>
<223>模板-校准物-alwni-n15
<220>
<221>misc_feature
<222>(44)..(58)
<223>n为a、c、g或t
<400>30
ggaagacgtagcaagggagtagccaatgtgaattcacacagtgnnnnnnnnnnnnnnnca60
ggggctggtcgtgactgggaaaaccctggc90
<210>31
<211>100
<212>dna
<213>人工序列
<220>
<223>标签-nabab-alwni
<220>
<221>misc_feature
<222>(32)..(39)
<223>n为a、c、g或t
<400>31
cacacaggaaacagctatgacccagtgtctgnnnnnnnntgtggacgctgtctcttatac60
acatctgacgctgccgacgacgtcgtgactgggaaaaccc100
<210>32
<211>20
<212>dna
<213>人工序列
<220>
<223>bg-m13-fwd**
<400>32
gggttttcccagtcacgacg20
<210>33
<211>22
<212>dna
<213>人工序列
<220>
<223>alexa488-m13-rev
<400>33
cacacaggaaacagctatgacc22
<210>34
<211>38
<212>dna
<213>人工序列
<220>
<223>特异于金黄色葡萄球菌的rnaiii的基于dna的探针
<400>34
gcagccagctggcctcgcttccgggatccgcgcagggg38
<210>35
<211>55
<212>dna
<213>人工序列
<220>
<223>特异于金黄色葡萄球菌的rnaiii的基于dna的探针
<220>
<221>misc_feature
<222>(36)..(41)
<223>n为a、c、g或t
<400>35
gatcgcggatcccggaagcgaggccagctggctgcnnnnnnttgggaggggctca55
<210>36
<211>62
<212>dna
<213>人工序列
<220>
<223>实体鉴定序列的实例
<220>
<221>misc_feature
<222>(34)..(43)
<223>n为a、c、g或t
<400>36
gagcggataacaatttcacacaggcacggggtgnnnnnnnnnngatcggaagagcgtcgt60
gt62
<210>37
<211>62
<212>dna
<213>人工序列
<220>
<223>实体鉴定序列的实例
<220>
<221>misc_feature
<222>(20)..(29)
<223>n为a、c、g或t
<400>37
acacgacgctcttccgatcnnnnnnnnnncaccccgtgcctgtgtgaaattgttatccgc60
tc62
- 还没有人留言评论。精彩留言会获得点赞!