酰胺键连接的聚合物的制备方法与流程
本发明涉及高分子化学领域,尤其涉及一种酰胺键连接的聚合物的制备方法。
背景技术:
嵌段聚合物在纳米器件、生物医药等领域有着非常好的应用前景,越来越多的科研工作者致力于嵌段聚合物的合成中。随着atrp等可控自由基聚合和点击化学等高效反应的发展,可以制备越来越多种类的嵌段聚合物,如对温度、光、ph、磁响应的嵌段聚合物。
目前嵌段聚合物的合成方法主要有活性阴离子聚合、活性自由基聚合、高效的点击反应和atrp、raft等可控自由基聚合结合的方法。其中阴离子聚合体系虽然简单,但反应条件非常的苛刻,体系对水和氧气比较敏感,通常要在高真空的条件下进行操作,因此使其应用受到了一定的限制。而atrp、raft等可控自由基方法聚合体系简单、单体适用范围较广,分子设计能力强的优点,但是对单体的种类仍然有一定的限制。而将点击化学反应如cuaac与atrp结合起来,可以打破其局限性,合成不同组成、结构和性能的嵌段聚合物。主要思路为用含有炔基的引发剂合成末端为炔基的聚合物,用普通引发剂合成末端为氯或溴的聚合物,在与叠氮化钠反应从而使聚合物末端叠氮化,最后将末端炔基和末端叠氮化的聚合物进行cuaac反应将两者连接起来,从而合成各种嵌段聚合物。
羧酸化合物与有机叠氮化物的反应被称为staudinger-vilarrasa反应,简称s-v反应。而目前催化的s-v反应主要被用到合成多肽以及含有酰胺键的天然大分子反应中。
技术实现要素:
为解决上述技术问题,本发明的目的是提供一种酰胺键连接的聚合物的制备方法,将二吡啶二硒醚催化的staudingers-vilarrasa反应用于聚合物之间的连接,通过含羧基化合物与一端为叠氮基团的聚合物之间的反应,形成了酰胺键,从而制备出酰胺键连接的聚合物。
本发明的酰胺键连接的聚合物的制备方法,包括以下步骤:
将一端为叠氮基团的聚合物、含羧基化合物、催化剂二吡啶二硒醚(pysesepy)混匀,在0-5℃下向其中加入三甲基膦(me3p)的有机溶液进行反应,当反应无气泡产生时,然后在25-40℃下反应2-24h,得到酰胺键连接的聚合物,
其中,含羧基化合物为二元羧酸、多元羧酸、一端为羧基的聚合物或两端为羧基的聚合物;
一端为叠氮基团的聚合物、二吡啶二硒醚、含羧基化合物和三甲基膦之间的摩尔比为0.9-2.2:1-2:1-2:20-40。
优选地,在0℃下加入三甲基膦(me3p)的有机溶液进行反应,当反应无气泡产生时,然后在40℃下反应。
当含羧基化合物为二元羧酸时,以上反应的路线如下:
式中,polymer-n3表示一端为叠氮基团的聚合物,hooc-r-cooh表示二元羧酸。
当含羧基化合物为多元羧酸时,以上反应的路线如下(以三元羧酸为例):
式中,polymer-n3表示一端为叠氮基团的聚合物。
当含羧基化合物为一端为羧基的聚合物时,以上反应的路线如下:
式中,polymer-n3表示一端为叠氮基团的聚合物,polymer1-cooh表示一端含羧基化合物。
当含羧基化合物为两端为羧基的聚合物时,以上反应的路线如下:
式中,polymer-n3表示一端为叠氮基团的聚合物,hooc-polymer2-cooh表示两端含羧基化合物。
进一步地,一端为叠氮基团的聚合物中的聚合物为聚乙二醇单甲醚、聚丙烯酸叔丁酯、聚苯乙烯等。
具体的,一端为叠氮基团的聚合物分别对应以下分子式:
叠氮化聚乙二醇单甲醚的制备方法如下:
将聚乙二醇单甲醚、三乙胺和溶剂混匀,冰浴条件下滴加对甲苯磺酰氯,然后在室温下反应24h,得到对甲苯磺酰氯基封端的聚乙二醇单甲醚。溶剂优选为二氯甲烷。
将对甲苯磺酰氯基封端的聚乙二醇单甲醚、叠氮化钠和溶剂混匀,在80℃反应48h,得到叠氮化聚乙二醇单甲醚,其中对甲苯磺酰氯基封端的聚乙二醇单甲醚和叠氮化钠之间的摩尔比为1:20。溶剂优选为n,n-二甲基甲酰胺。
叠氮化聚丙烯酸叔丁酯的制备方法如下:
将聚丙烯酸叔丁酯、叠氮化钠和溶剂混匀,于60℃反应24h,得到叠氮化聚丙烯酸叔丁酯,其中聚丙烯酸叔丁酯和叠氮化钠之间的摩尔比为1:10;溶剂优选为n,n-二甲基甲酰胺。
在制备一端为叠氮基团的聚合物过程中采用了atrp方法以及末端为羟基的聚合物和对甲苯磺酰氯反应,最后在与叠氮化钠亲核反应,简单方便,适合聚合物的范围广。
进一步地,一端为叠氮基团的聚合物的分子量为2000-5200da。
进一步地,含羧基化合物为二元羧酸或多元羧酸,一端为叠氮基团的聚合物、二吡啶二硒醚、含羧基化合物和三甲基膦之间的摩尔比为1.9-2.2:2:1:20-40。
进一步地,含羧基化合物为对苯二甲酸、十四烷二酸、均苯三酸等。
进一步地,含羧基化合物为一端为羧基的聚合物或两端为羧基的聚合物,一端为叠氮基团的聚合物、二吡啶二硒醚、含羧基化合物和三甲基膦之间的摩尔比为0.9-1.3:1:2:20-40。
进一步地,一端为羧基的聚合物或两端为羧基的聚合物中的聚合物为聚乙二醇、聚乙二醇单甲醚等。
具体的,一端为羧基的聚合物对应以下分子式:
具体的,两端为羧基的聚合物对应以下分子式:
进一步地,含羧基化合物为一端为羧基的聚合物或两端为羧基的聚合物时,其制备方法如下:
将末端为羟基的聚合物、丁二酸酐、4-二甲氨基吡啶、三乙胺和溶剂混匀,在25℃下通气状态下搅拌反应24-48h,得到末端羧基的聚合物,其中末端为羟基的聚合物、丁二酸酐、4-二甲氨基吡啶和三乙胺的摩尔比为1:1.5-3:1.5-3:1.5-3。
进一步地,三甲基膦的有机溶液中的有机溶剂为甲苯。使用甲苯作为溶剂,反应效率最高。
进一步地,三甲基膦的有机溶液的浓度为1mol/l。
进一步地,反应均在惰性气体保护下进行。具体是将惰性气体通入反应溶液中。
进一步地,惰性气体为氮气、氦气或氖气中的任意一种,优选氮气。
进一步地,催化剂二吡啶二硒醚的制备方法如下:
将氢氧化钠、硒粉、水合肼溶于有机溶剂,在25℃下反应2h,向其中加入2-溴吡啶,然后在120℃下反应24h,得到催化剂二吡啶二硒醚。
进一步地,氢氧化钠、硒粉、水合肼和2-溴吡啶之间的摩尔比为1.5:1:1:1。
进一步地,有机溶剂为n,n’-二甲基甲酰胺。
进一步地,水合肼的浓度为85%。
在本发明的制备方法中,加入三甲基膦的有机溶液后,迅速与末端叠氮化的聚合物反应生成膦腈,由于三甲基膦是过量的,并且生成膦腈的反应可以在几分钟内完成,所以能快速地把全部的叠氮基团转化为膦腈,同时二吡啶二硒醚与部分含羧基化合物反应生成活性酯,膦腈与活性酯迅速反应生中间体,且由于二吡啶二硒醚与含羧基化合物的摩尔比为1-2:1-2,二吡啶二硒醚只能活化部分的含羧基化合物,剩余的含羧基化合物与中间体反应得到目标产物以及活性酯,再以此循环下去直至底物反应完全。以二元羧酸为例,反应原理如下:
借由上述方案,本发明至少具有以下优点:
(1)本发明首次将atrp和二吡啶二硒醚催化的staudinger-vilarrasa反应结合起来,选用atrp和亲核取代反应制备的一端为叠氮基团的聚合物,然后利用s-v反应,将含羧基化合物与其依靠酰胺键连接起来,为聚合物的相互连接提供了一种新方法。
(2)本发明在酰胺化时采用了二吡啶二硒醚催化的staudinger-vilarrasa反应,具有适用范围广、反应速率快、产率高、副产物少、易处理等显著特点,是一种高效的连接聚合物的方法。
上述说明仅是本发明技术方案的概述,为了能够更清楚了解本发明的技术手段,并可依照说明书的内容予以实施,以下以本发明的较佳实施例并配合附图详细说明如后。
附图说明
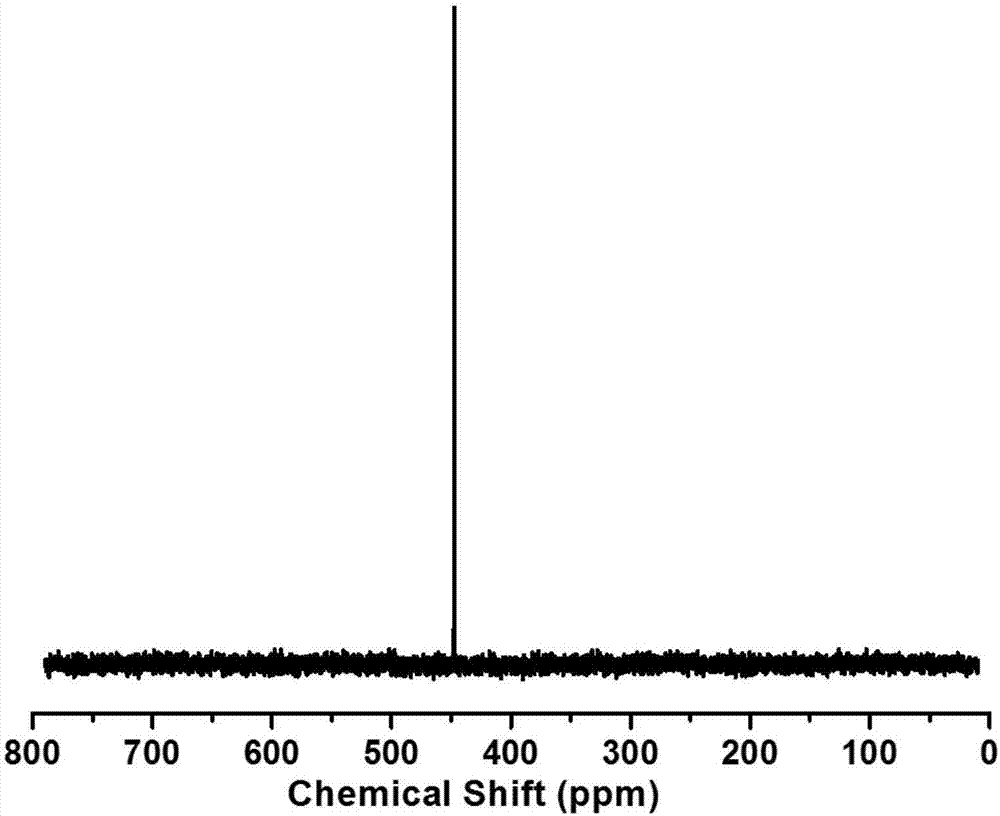
图1为实施例1制备的二吡啶二硒醚的核磁共振氢谱;
图2为实施例1制备的二吡啶二硒醚的核磁共振硒谱;
图3为实施例1制备的peg1900-n3的核磁共振氢谱;
图4为实施例1制备的peg1900-n3的大分子质谱;
图5为实施例1中peg1900-n3以及其与对苯二甲酸连接后的产物的核磁共振氢谱;
图6为实施例1中peg1900-n3以及其与对苯二甲酸连接后的产物的gpc流出曲线;
图7为实施例2中peg1900-n3与均苯三酸连接后的产物的核磁共振氢谱;
图8为实施例2中peg1900-n3与均苯三酸连接后的产物的gpc流出曲线;
图9为实施例3制备的ptba-n3的gpc流出曲线图;
图10为实施例3制备的ptba-n3的核磁共振氢谱;
图11为实施例3制备的peg1900-cooh的核磁共振氢谱;
图12为实施例3制备的peg1900-cooh及原料peg1900-oh的红外光谱图;
图13为实施例3制备的peg1900-cooh的大分子质谱图;
图14为实施例3中peg1900-cooh、ptba-n3、peg-b-ptba的gpc流出曲线图;
图15为实施例4制备的hooc-peg2050-cooh的核磁共振氢谱;
图16为实施例4制备的hooc-peg2050-cooh的大分子质谱图;
图17为实施例4制备的ptba-b-peg-b-ptba的核磁共振氢谱;
图18为实施例4中ptba-n3、ptba-b-peg-b-ptba的gpc流出曲线图。
具体实施方式
下面结合附图和实施例,对本发明的具体实施方式作进一步详细描述。以下实施例用于说明本发明,但不用来限制本发明的范围。
本发明以下实施例中采用以下测试仪器及条件:
凝胶渗透色谱(gpc):使用waters1515凝胶色谱仪测定,使用示差折光检测器(ri2414),hr1、hr2和hr4柱子的分子量范围为100-500000da,以thf为流动相,流速为1.0ml/min,在30℃下进行测试,用聚苯乙烯标样或者聚丙烯酸甲酯标样对聚合物分子量进行标定;
核磁共振氢谱(1h-nmr):使用bruker300mhz核磁仪,以cdcl3(氘代氯仿)为溶剂,tms(四甲基硅烷)为内标,室温下测定;
核磁共振硒谱(77se-nmr):使用bruker600mhz核磁仪,以cdcl3为溶剂,tms为内标,室温下测定;
基质辅助激光解析电离飞行时间质谱(maldi-tof):使用brukerultraflex-iiimsspectrometer质谱仪,以dctb为基质;
元素分析(ea):使用ea1110-chno-s微量分析仪测定;
红外光谱(ft-ir)采用brukertensor-27红外光谱仪测定,kbr压片法。
实施例1
(1)将6.1g(152mmol)的氢氧化钠、7.9g(100mmol)的硒粉和250ml的n,n’-二甲基甲酰胺溶剂加入到500ml的三颈烧瓶中,在通气状态下搅拌,再将加入4.9g的水合肼慢慢加入到体系中,然后在室温下反应2h。随后再将2-溴吡啶15.8g(100mmol)缓慢滴加到反应瓶中,然后升温到120℃下反应24h,停止反应后恢复到室温。抽滤,滤液用大量乙酸乙酯进行萃取。有机相再用分别用饱和氯化铵溶液、去离子水饱和食盐水各洗一次,再用无水硫酸钠搅拌干燥2h,浓缩得粗产品,再用柱层析进行提纯。最终得到9.7g最终产物pysesepy,产率为62%。以上反应的路线如下:
对产物分别进行核磁共振氢谱(图1)、核磁共振硒谱(图2)和元素分析测试(表1),图1中各峰都能找到归属,并且积分正确,图2为单峰,表明合成的为单硒醚或二硒醚,再结合表1中元素分析的结果,表明成功合成了二吡啶二硒醚pysesepy。
表1pysesepy的元素分析测试结果
(2)将15g(7.8947mmol)聚乙二醇单甲醚(命名为peg1900-oh,分子量为1900da)、15.9774g(0.1579mmol)三乙胺和200ml的二氯甲烷加入到500ml的圆底烧瓶中,反应体系放入冰盐浴中搅拌,用150ml的二氯甲烷将30.10g(157.9mmol)的对甲苯磺酰氯溶解,然后用恒压滴液漏斗将对甲苯磺酰氯的溶液滴加到反应体系中,滴加完毕后,移去冰浴,在室温下搅拌反应24h。反应停止后抽滤除去生成的盐,浓缩滤液,然后沉淀在冷的无水乙醚中,静置,抽滤,真空干燥,最后得到14g的对甲苯磺酰氯基封端的聚乙二醇单甲醚(命名为peg1900-ots)。将14g(7.0mmol)的peg1900-ots、4.55g(70mmol)的叠氮化钠和60ml的n,n-二甲基甲酰胺加入到100ml的圆底烧瓶中,在80℃反应24h,再用中性氧化铝柱除去各种钠盐,沉淀在冷的无水乙醚中,抽滤,真空干燥,最后得到6.9g的末端叠氮化的聚乙二醇单甲醚(命名为peg1900-n3)。其反应路线如下:
图3是peg1900-n3的核磁共振氢谱,由核磁计算得到的数均分子量mn=2400da,各峰都能找到对应的归属,并且积分正确。图4是peg1900-n3的大分子质谱,图中存在三组峰,第一组峰归属于[peg-n3+na]+(实验值为1841.01与理论值1841.21,n=39相一致)这一离子碎片。而其余两组峰归属于[peg-n+na]+(实验值为1857.08与理论值1857.24,n=40相一致)和亚稳离子(实验值为1860.11),这两个离子碎片都是由于在大分子质谱测试过程中聚合物末端叠氮基团释放n2形成的。用凝胶渗透色谱gpc测得其数均分子量mn=2600da,分子量分布pdi=1.14(图6中a曲线)。
(3)在50ml的反应管中,加0.4800g(0.24mmol)的末端叠氮化的聚乙二醇单甲醚peg1900-n3,0.0754g(0.024mmol)的二吡啶二硒醚pysesepy和0.0202g(0.12mmol)的对苯二甲酸,搅拌并通入惰性气体,再将温度为0℃的三甲基膦的甲苯溶液4.0ml缓慢注入到体系中,当体系中无气体产生时,将体系温度升到40℃,通气状态下继续反应24h。反应停止后加入适量的四氢呋喃稀释,再滴加到冷的无水乙醚中进行沉淀,静置,抽滤,放入真空箱中干燥,最终得到0.4370g的用对苯二甲酸偶联在一起的聚乙二醇单甲醚(peg-peg),收率为89.3%。反应路线如下:
图5为对苯二甲酸与末端叠氮化的聚乙二醇单甲醚peg1900-n3偶联之后的核磁氢谱,与偶联之前相比,在7.86-7.89ppm处出现了对苯二甲酸苯环上质子氢(d)的特征峰,表明偶联反应的成功,由图5中特征峰(d)和特征峰(b)可以计算出偶联效率可达93.9%。图6为连接前聚乙二醇单甲醚(曲线a)以及其与对苯二甲酸偶联后的gpc流出曲线(曲线b),图中b曲线中,编号为2的峰的数均分子量为mn=5100da,分子量分布pdi=1.02,编号为1的峰数均分子量mn=2500da,分子量分布pdi=1.03,其中编号为2的峰是偶联产物(peg-peg)的出峰,通过高斯函数可以计算出偶联产物所占的比率为91.5%。测试的结果表明对苯二甲酸成功将末端叠氮化的聚乙二醇单甲醚peg1900-n3偶联起来。
通过以上分析可知,在对苯二甲酸与聚乙二醇单甲醚peg1900-n3偶联反应中,催化的staudinger-vilarrasa反应的偶联效率可以达到93.9%。
实施例2
(1)在50ml的反应管中,加0.3840g(0.192mmol)的实施例1中制备的末端叠氮化的聚乙二醇单甲醚peg1900-n3,0.0565g(0.18mmol)的二吡啶二硒醚pysesepy和0.0128g(0.06mmol)的均苯三酸,搅拌并通入惰性气体,再将温度为0℃的三甲基膦的甲苯溶液2.8ml缓慢注入到体系中,当体系中无气体产生时,将体系温度升到40℃,通气状态下继续反应24h。反应停止后加入适量的四氢呋喃稀释,再滴加到冷的无水乙醚中进行沉淀,静置,抽滤,放入真空箱中干燥,最终得到0.3875g的用均苯三酸偶联末端叠氮化的聚乙二醇单甲醚形成的三臂聚合物,收率为99.0%。反应路线如下:
图7为均苯三酸与末端叠氮化的聚乙二醇单甲醚peg1900-n3偶联之后的核磁氢谱,与偶联之前相比,在8.48-8.50ppm处出现了均苯三酸苯环上质子氢(a)的特征峰,表明反应的成功,由图7中特征峰(a)和特征峰(c)可以计算出偶联效率可达94.9%。图8为反应前聚乙二醇单甲醚(曲线a)以及其与均苯三酸反应后的gpc流出曲线(曲线b),图中b曲线中,编号为2的峰的数均分子量为mn=7200da,分子量分布pdi=1.02,编号为2的峰数均分子量mn=2600da,分子量分布pdi=1.02,其中编号为2的峰是反应产物的出峰,通过高斯函数可以计算出偶联产物所占的比率为95.0%。测试的结果表明均苯三酸成功将末端叠氮化的聚乙二醇单甲醚peg1900-n3连接起来,形成了三臂星形聚合物。
通过以上分析可知,在均苯三酸与末端叠氮化的聚乙二醇单甲醚peg1900-n3反应中,催化的staudinger-vilarrasa反应的连接效率可以达到95.0%。
实施例3
(1)将18.88g(147.3mmol)的丙烯酸叔丁酯、0.3175g(2.2130mmol)的cubr、0.0247g(0.1106mmol)的cubr2、0.4026g(2.3230mmol)的pmdeta(n,n,n’,n’,n”-五甲基二乙烯三胺)、0.7391g(4.4260mmol)的2-溴丙酸甲酯(mbrp)和20ml的二甲亚砜加入到50ml的schlenk瓶中,随后在抽气-充气3次,放入到60℃的油浴锅中反应3h。将反应淬灭后,用四氢呋喃把产物溶解后,通过中性氧化铝柱除去铜盐,沉淀在甲醇和水的混合溶剂中,抽滤,真空箱干燥24h,最终得到11.5g的聚丙烯酸叔丁酯(命名为ptba-br),收率为58.6%,再用凝胶渗透色谱gpc测得的数均分子量mn=5000da,分子量分布pdi=1.17。
(2)将6g(1.2mmol)的ptba-br、1.5602g(24mmol)的nan3和30ml的dmf加入到50ml的圆底烧瓶中,在60℃的油浴锅中搅拌反应24h。停止反应恢复到室温后,加入适量的四氢呋喃溶解后,再用中性氧化铝柱除去各种钠盐,沉淀在甲醇和水中,静置,抽滤,放入真空箱干燥过夜,最后得到4.16g的白色粉末状的叠氮化聚丙烯酸叔丁酯(ptba-n3),收率为69.3%,再用凝胶渗透色谱gpc测得的数均分子量mn=5200da,分子量分布pdi=1.15,其gpc流出曲线如图9所示。图10为其核磁共振氢谱,由核磁计算得到的分子量mn=4600da,图中各峰都能找到对应的归属,并且积分正确,由此可知,叠氮化产物ptba-n3成功合成。步骤(1)-(2)的合成路线如下:
(3)将10g(5.2632mmol)的聚乙二醇单甲醚peg1900-oh(分子量为1900),0.7900g(7.8947mmol)的丁二酸酐sa,0.9645g(7.8947mmol)的4-二甲氨基吡啶(dmap)、0.7989g(7.8947mmol)的三乙胺和400ml的1,4-二氧六环加入到500ml的三颈烧瓶中,搅拌并通气,在25℃的条件下反应24h。反应停止后先抽滤除去析出的盐,滤液浓缩,在沉淀到冷的无水乙醚中,静置,抽滤,放入真空箱中干燥,最终得到9g的末端羧基化的聚乙二醇单甲醚(命名为peg1900-cooh),收率为90%。反应路线如下:
图11为peg1900-cooh的核磁共振氢谱,图中各峰都能找到对应的归属,并且积分正确,由核磁计算得到的分子量mn=2300da。图12为酯化反应前后红外光谱图,a曲线代表产物peg1900-cooh,b曲线代表原料peg1900-oh,由图可知反应之后在a曲线的1732cm-1出现羰基的特征峰,说明酯化反应的成功。图13为合成的peg1900-cooh大分子质谱图,图中存在两组峰分布,第一组峰归属于[peg-cooh+h]+(实验值为2070.24与理论值2070.36,n=43相一致)这一离子碎片。另外一组峰归属于[peg-cooh+na]+(实验值为2092.32与理论值2092.35,n=43相一致),即各峰都能找到对应的归属,并且实验值和理论值的误差在允许的范围之内。图11、图12和图13都很好的证明了成功合成了末端羧基化的聚乙二醇单甲醚peg1900-cooh。
(4)在50ml的反应管中,加入0.2080g(0.04mmol)的ptba-n3,0.0800g(0.04mmol)的peg1900-cooh,和0.0126g(0.04mmol)二吡啶二硒醚pysesepy,搅拌并通入惰性气体,再将温度为0℃的三甲基膦的甲苯溶液1.5ml缓慢注入到体系中,当体系中无气体产生时,将体系温度升到40℃,通气状态下继续反应24h。反应停止后加入适量的四氢呋喃稀释,再滴加到冷的无水乙醚中进行沉淀,静置,抽滤,放入真空箱中干燥,最终得到0.2359g的两嵌段聚合物peg-b-ptba,收率为82.4%。反应路线如下:
图14为连接前后的gpc流出曲线,其中,a曲线代表peg1900-cooh(数均分子量mn=2200da,分子量分布pdi=1.07),b曲线代表ptba-n3(数均分子量mn=5200da,分子量分布pdi=1.15),c曲线代表嵌段聚合物peg-b-ptba(数均分子量mn=7500da,分子量分布pdi=1.17)。测试的结果表明末端羧基化的聚乙二醇单甲醚peg1900-cooh和末端叠氮化的聚丙烯酸叔丁酯ptba-n3成功连接起来,连接的效率可达100%。
实施例4
(1)将10g(4.8780mmol)的聚乙二醇ho-peg2050-oh(分子量为2050),1.4644g(14.63mmol)的丁二酸酐sa,1.7873g(14.63mmol)的4-二甲氨基吡啶(dmap)、1.4804g(14.63mmol)的三乙胺和400ml的1,4-二氧六环加入到500ml的三颈烧瓶中,搅拌并通气,在25℃的条件下反应24h。反应停止后,先抽滤除去析出的盐,滤液浓缩,再沉淀到冷的无水乙醚中,静置,抽滤,放入真空箱中干燥,最终得到8.8g的末端羧基化的聚乙二醇hooc-peg2050-cooh,收率为88.0%。反应路线如下:
图15为hooc-peg2050-cooh的核磁共振氢谱,图中各峰都能找到对应的归属,并且积分正确,由核磁计算得到的分子量mn=2300da。图16为合成的hooc-peg2050-cooh大分子质谱图,图中只有一组峰,归属于[hooc-peg-cooh+na]+,实验值(2310.32)与理论值(2310.44,n=45)的误差在实验允许的范围之内。图15和图16都很好的证明了成功合成了末端羧基化的聚乙二醇hooc-peg2050-cooh。
(2)在50ml的反应管中,加入0.2600g(0.05mmol)的实施例2制备的ptba-n3,0.0575g(0.025mmol)的hooc-peg2050-cooh和0.0157g(0.05mmol)二吡啶二硒醚pysesepy,搅拌并通入惰性气体,再将温度为0℃的三甲基膦的甲苯溶液1.5ml缓慢注入到体系中,当体系中无气体产生时,将体系温度升到40℃,通气状态下继续反应24h。反应停止后加入适量的四氢呋喃稀释,再滴加到冷的无水乙醚中进行沉淀,静置,抽滤,放入真空箱中干燥,最终得到0.2661g的三嵌段聚合物ptba-b-peg-b-ptba,收率为85.0%。反应路线如下:
图17为连接之后的三嵌段聚合物ptba-b-peg-b-ptba的核磁共振氢谱图,由核磁计算得到的分子量mn=14200da。图18为连接前后的gpc流出曲线,其中,a曲线代表ptba-n3(数均分子量mn=5200da,分子量分布pdi=1.15),b曲线代表ptba-b-peg-b-ptba(数均分子量mn=14400da,分子量分布pdi=1.11)。测试的结果表明末端羧基化的聚乙二醇hooc-peg2050-cooh和末端叠氮化的聚丙烯酸叔丁酯ptba-n3成功地连接起来,生成了三嵌段聚合物ptba-b-peg-b-ptba。
以上所述仅是本发明的优选实施方式,并不用于限制本发明,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和变型,这些改进和变型也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!