一种氧杂螺环PNN类型配体的合成与应用的制作方法
本发明涉及一种新型氧杂螺环双膦配体的合成。该化合物可以作为手性配体应用于不对称催化反应中,在不对称催化领域具有很高的潜在应用价值,属于不对称催化领域。
背景技术:
1987年,noyori用binap/ru(ococh3)2/hcl引发了对官能化酮不对称氢化的革命,其后,他发展的手性双膦/钌/双胺催化剂将底物范围拓展到了非官能化酮((a)noyorir.etal.j.am.chem.soc.1987,109,5856;(b)ohkumat.etal.j.am.chem.soc.1995,117,2675)。但是,其催化体系还有很多缺陷,比如说某些官能化酮可以取代双胺配体安耐吉使催化体系失去活性,致使底物范围受限;再比如说手性双膦配体和手性双胺配体的立体效应必须精确匹配才能取得很高的对映选择性的结果。所以合成一种既带有手性膦/胺配体特征,结构又可以简单的配体是潜力巨大的一个研究发展方向。
现在大部分的催化氢化优势配体是轴手性双齿配体,但三齿配体理论上可以形成更精准更稳定的手性立体结构。因此,轴手性sp3p/sp3n/sp2n线性三齿形式配体潜力巨大。2011年,周其林教授设计并合成了spiropap配体,其在α,β-不饱和酮和简单芳基烷基酮的催化上取得了不错的成果(xiej.;liux.;xiej.;wangl.;zhouq.angew.chem.int.ed.2011,50,7329)。2013年,yamamura设计了binan-py-pph2,完成了对叔烷基底物的攻克(yamamurat.;nakatsukah.;tanakas.;kitamuram.angew.chem.int.ed.2013,52,9313)。2016年,张绪穆教授成功完成了f-amphox配体,其合成更加简单,催化剂转换数更高(wuw.;lius.;duanm.;tanx.;chenc.;xiey.;lany.;dongx.;zhangx.organicletters,2016,18,2938)。
这种配体现在面临的难题是其在八面体金属/配体几何模型中有facial和meridional两种构象异构,fac类型可以进行分子间催化氢化,mer类型可以进行分子内催化氢化,所以具有单一的构象才能实现高效不对称催化氢化,形成单一的构象的解决办法有两种:放弃其线性结构或者增加结构的刚性(yamamurat.;nakanes.;nomuray.;tanakas.;kitamuram.,tetrahedron,2016,72,3781-3789)。此次研发的催化剂与之前的催化剂相比,具有刚性更大,合成步骤更简单的优势,所以具有非常大的研究与应用价值。
技术实现要素:
鉴于现有技术需要改善的问题,本发明提供了一种氧杂螺环pnn类型配体,具有以下通式(i)的结构:
通式(i)中:
r1、r2独立为烷基、烷氧基、芳基、芳氧基或者氢原子,r1、r2、r3和r4可成环或不成环;r5、r6独立为烷基、芳基或者氢原子;r7、r8为烷基、苄基或者芳基。
上述术语烷基优选为甲基、乙基、丙基、丁基等;
烷氧基优选为甲氧基、乙氧基、丙氧基、丁氧基等;
芳基优选为被烷基或者烷氧基取代或者未取代的苯基等,烷基和烷氧基如上定义。
所述一种氧杂螺环pnn类型配体为(±)-氧杂螺环双膦配体,(+)-氧杂螺环双膦配体,或(-)-氧杂螺环双膦配体。
优选的方案之一为:所述氧杂螺环pnn类型配体具有下式(6)的结构:
一种合成前述化合物的方法,通过下面的路线合成得到:
本发明另一目的在于提供前述化合物在催化不对称反应中的应用,所述不对称反应包括氢化反应,氢甲酰化反应,硅氢化反应,硼氢化反应,氢羟基化反应,氢氨化反应,氢氰基化反应,异构化甲酰基化反应,氢氨甲基化反应,转移氢化反应,烯丙基化反应,烯烃复分解反应,环异构化反应,diels-alder反应,不对称偶联反应,aldol反应,michael加成反应,不对称环氧化反应,动力学拆分和[m+n]环化反应。
根据前述的应用,该化合物制成的双膦醋酸钌络合物在有机溶剂中对不饱和羧酸的氢化具有很高的活性和对映选择性,大于99%。
优选前述应用的合成路线如下:
本发明的另一目的在于提供一种催化不对称反应,使用权前述的化合物作为催化剂,反应路线如下:
本发明相对于现有技术的有益效果包括:
(1)该氧杂螺环pnn类型配体化合物具有中心手性,因此有左旋氧杂螺环pnn类型配体和右旋氧杂螺环pnn类型配体,消旋的氧杂螺环pnn类型配体可以通过消旋的氧杂螺环二酚为原料合成得到。
(2)本发明可以作为手性配体应用于不对称氢化当中,该化合物制成的金属铱的络合物在有机溶剂中对轴手性内酯的氢化具有很高的活性和对映选择性,大于99%。同时该催化剂在简单酮的氢化中可以得到98%的对映选择性。
附图说明
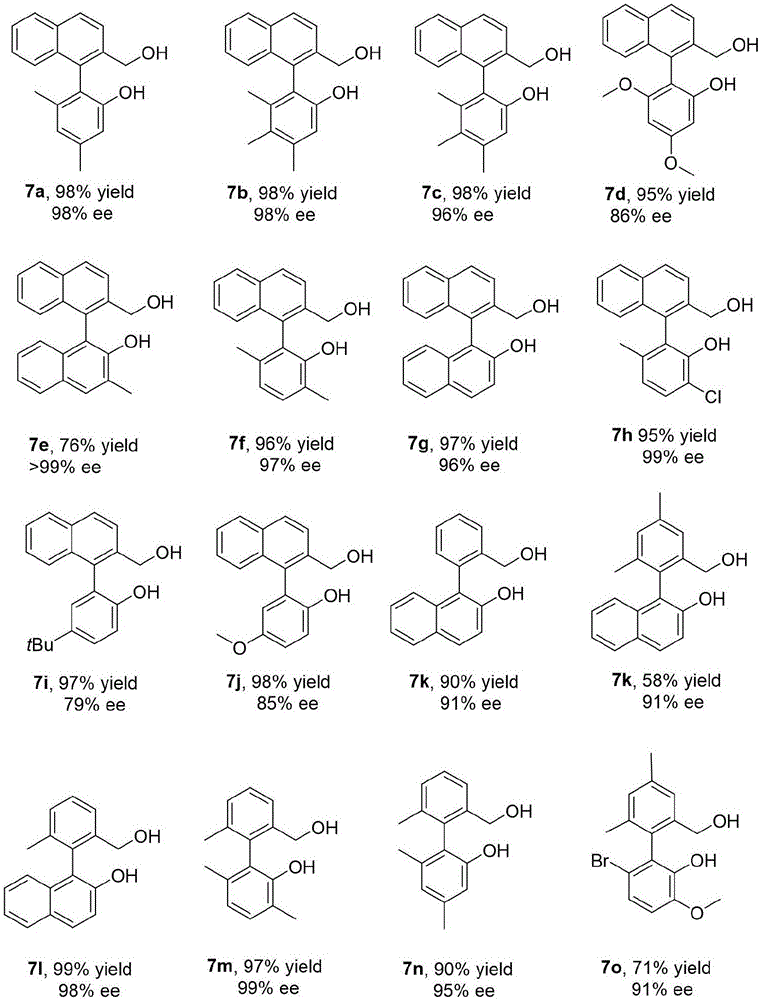
图1,本发明多种不同手性化合物的制备示意图,以及对应的转化率和对映选择性ee值。
具体实施方式
下面通过实施例和附图对本发明加以说明,但本发明并不仅限于以下实施例。
实施例1:
(s)-4'-(二3,5-二叔丁基基苯基膦氧基)-2氢,2'氢-3,3'-螺二[苯并呋喃]-4-三氟甲磺酸酯2a的合成:
n2氛围下,反应瓶中加入1(5.2g,10mmol)、dppb(213mg,0.05mmol)、ar2poh(6.39g,15mmol)、pd(pac)2(112mg,0.05mmol)和dipea(6.5ml,40mmol),最后加入50ml无水无氧的dmso。100℃下反应6h。冷却至室温后,加入水淬灭反应,用乙酸乙酯萃取反应体系,有机相用无水硫酸钠干燥后减压条件下除去溶剂,经过简单的柱层析后就可以拿到产物2a(7.43g,产率=93%)。
白色固体.1hnmr(500mhz,cdcl3)δ1.18(s,9h,ch3),1.19(s,9h,ch3),1.20(s,9h,ch3),1.21(s,9h,ch3),4.54-4.56(m,1h,ch2),4.62-4.64(m,1h,ch2),4.80-4.82(m,2h,ch2),6.63-6.68(m,2h,ar),6.82-6.88(m,6h,ar),6.90-6.94(m,2h,ar),7.15-7.21(m,1h,ar),7.26-7.34(m,1h,ar).13c{1h}nmr(126mhz,cdcl3)δ162.9,160.1,150.5,150.2,145.7,136.0(m),135.5,134.4,131.2,129.6,128.4,127.5,122.4(m),112.5,110.5,109.6,83.5,82.3,56.4(m),34.8(m),31.3(m).31p{1h}nmr(202mhz,cdcl3)δ-19.74(s).hrms(esi)计算值:c44h53o6f3ps[m+h]+:797.3253,实验值:797.3247,[α]20d=-109.6(c=0.5,丙酮).
实施例2:
(s)-4'-(二3,5-二叔丁基苯基膦基)-2氢,2'氢-3,3'-螺二[苯并呋喃]-4-三氟甲磺酸酯3a的合成:
100ml封管中,加入2a(3.98g,5mmol)、dipea(6.6ml,40mmol)、20ml甲苯和三氯硅氢(2.0ml,20mmol)。反应在120℃条件下搅拌过夜。反应体系用过量的碳酸氢钠饱和溶液淬灭,加入100ml乙酸乙酯,硅藻土过滤,有机相用无水硫酸钠干燥。减压下除去溶剂,然后柱层析得到白色固体3a(3.51g,产率=90%)。
白色固体.1hnmr(500mhz,cdcl3)δ1.17(s,9h,ch3),1.19(s,9h,ch3),1.20(s,9h,ch3),1.21(s,9h,ch3),4.52-4.55(m,1h,ch2),4.60-4.63(m,1h,ch2),4.77-4.82(m,2h,ch2),6.61-6.69(m,2h,ar),6.80-6.94(m,6h,ar),7.14-7.22(m,2h,ar),7.28-7.34(m,2h,ar).13c{1h}nmr(126mhz,cdcl3)δ162.9,160.1(m),150.5(m),145.8,136.1(m),135.5(m),134.4,131.9(m),131.2,129.6,128.4(m),127.5,122.4(m),116.9,112.6,110.6,109.6,83.5,82.4,56.4(m),34.8(m),31.3(m).31p{1h}nmr(202mhz,cdcl3)δ-19.73(s).hrms(esi)计算值:c32h29o5f3ps[m+h]+:781.3303,实验值:781.3298,[α]20d=-78.8(c=0.5,丙酮).
实施例3:
(s)-4-二3,5-二叔丁基苯基磷基-2h,2'h-3,3'-螺二苯并四氢呋喃-4-甲酸4a的制备:
向一个250ml的反应釜中加入醋酸钯(259mg,1.16mmol)和1,3-双(二苯基膦)丙烷(478mg,1.16mmol),3a(5.51g,7.0mmol)。然后将反应釜转移到手套箱中,依次加入二甲亚砜(60ml)和甲醇(42ml)和三乙胺(11.6ml)。然后将反应釜移出手套箱,将一氧化碳的压力加至10atm。然后将反应釜放置于70℃油浴上搅拌过夜。反应进行完全后,将反应混合液转移到500ml分液漏斗中加入250ml乙酸乙酯,然后用饱和食盐水洗涤有机相两次,将有机相干燥,除去溶剂柱层析得到甲酯产物,然后经过水解就可以得到目标产物4a(2.84g,4.2mmol,产率=60%)
白色固体.1hnmr(400mhz,cdcl3)δ7.36–7.17(m,4h),7.17–7.00(m,2h),6.94(d,j=6.5hz,1h),6.76(t,j=8.5hz,4h),6.54(s,1h),4.98(d,j=8.4hz,1h),4.74(d,j=8.3hz,2h),4.63(d,j=8.5hz,1h),1.16(s,18h),1.13(s,18h).13c{1h}nmr(101mhz,cdcl3)δ169.65,162.27,160.82,160.71,150.12,150.05,150.00,149.93,136.44,136.36,135.30,135.04,134.13,133.86,133.67,132.28,132.25,129.35,128.60,127.75,127.66,127.56,127.45,126.94,126.76,123.96,122.58,121.83,114.62,109.73,85.24,83.42,58.06,58.02,34.71,34.64,31.27,31.23.31p{1h}nmr(162mhz,cdcl3)δ-20.01(s).hrms(esi)计算值:c44h54o4p[m-h]-:677.3754,实验值:677.3745,[α]20d=-32.0(c=0.5,丙酮).
实施例4:
(s)-4-二苯基磷基-4’-羧基-3.3’-螺二苯并四氢呋喃的制备:
氩气保护下,0℃条件下向一个100ml反应瓶中加入4a(676mg,1mmol)、甲苯(10ml),dppa(303mg,1.1mmol)、三乙胺(152mg,1.5mmol)。加入完毕后搅拌20min,然后将温度升至90℃继续反应30min。然后加入2mlmeoh,继续反应30min,最后加入4.0m的koh溶液(4ml),回流条件下搅拌过夜。反应体系用二氯甲烷萃取(30ml*2),干燥后,减压条件下除去溶剂,柱层析可以得到产物5a(350mg,产率=54%)。
白色固体.1hnmr(400mhz,cdcl3)δ7.40-7.26(m,2h,ar),7.20(t,j=7.8hz,1h,ar),7.00–6.76(m,6h,ar),6.67(dd,j=7.6,4.0hz,1h,ar),6.30(d,j=7.9hz,1h,ar),5.70(d,j=7.9hz,1h,ar),5.05(dd,j=9.1,2.4hz,1h,ch2),4.65(dd,j=9.1,3.8hz,2h,ch2),4.56(d,j=9.3hz,1h,ch2),2.85(s,2h,nh2),1.20(s,18h,ch3),1.17(s,18h,ch3).13c{1h}nmr(101mhz,cdcl3)δ161.3,160.2,150.2,150.2,150.1,150.0,143.6,136.6,136.1,134.2,130.42,129.5,128.0,127.9,127.7,122.7,121.9,113.9,110.6,108.8,100.34,8.16,80.5,56.2,34.79,34.8,31.4,31.3.31p{1h}nmr(162mhz,cdcl3)δ-20.66(s).hrms(esi)计算值:c43h55no2p[m+h]+:648.3970,实验值:648.3973,[α]20d=-80.6(c=0.5,丙酮).
实施例5:
(s)-4-二苯基磷基-4’-氨基-3.3’-螺二苯并四氢呋喃的制备:
在氩气保护下,向一个10ml反应瓶中依次加入5a(194mg,0.3mmol)、3-甲基吡啶-2-甲醛(45.4mg,0.37mmol)、醋酸硼氢化钠(211mg,1mmol)以及dce(3ml),室温下搅拌过夜。过滤然后减压下除去溶剂,柱层析得到目标产物6a(185mg,产率=82%)。
白色固体.1hnmr(500mhz,cdcl3)δ7.93-7.94(m,1h,ar),7.31-7.33(m,1h,ar),7.24-7.27(m,3h,ar),7.09-7.17(m,1h,ar),6.94-6.99(m,2h,ar),6.86-6.88(m,2h,ar),6.74-6.76(m,3h,ar),6.32(d,j=8.0hz,1h,ar),6.04(d,j=8.0hz,1h,ar),5.66-5.67(br,1h,nh2),4.69-4.71(m,1h,ch2),4.57-4.64(m,3h,ch2),4.11(dd,j1=5.5hz,j2=16.0hz,1h,ch2),3.59(d,j=15.5hz,1h,ch2),2.15(s,3h,ch3),1.17(s,18h,ch3),0.96(s,18h,ch3).13c{1h}nmr(126mhz,cdcl3)δ161.3,160.7(m),153.8,150.1(m),149.7,145.6,144.9,136.9,135.0,131.6(m),130.7,129.8,129.0,128.0(m),121.9(m),121.5,111.6,110.4,103.3,99.0,83.2,80.4,56.4,44.8,34.5(m),31.1(m),17.1.31p{1h}nmr(202mhz,cdcl3)δ-19.44(s).hrms(esi)计算值:c50h62o2n2p[m+h]+:753.4543,实验值:753.4517,[α]20d=-117.8(c=0.5,丙酮).
实施例6:
配体6a和[ir(cod)cl]2原位催化苯乙酮的不对称氢化反应:
在氩气氛围的手套箱内,配体l1(7.9mg,0.0105mmol)和[ir(cod)cl]2(3.4mg,0.005mmol)加入2.5ml小瓶中,用etoh(1.0ml)溶解后室温络合1h得催化剂溶液。取催化剂溶液(20ul,0.0002mmol)加入5ml的氢化小瓶中,然后依次加入1.6mgtbuoli(0.02mmol),底物苯乙酮(0.23ml,2mmol),2ml异丙醇溶剂。将氢化小瓶放入氢化反应釜中,氢气置换三次后充入20atmh2,在室温下反应6h。反应完毕释放氢气后,反应混合物过一个短的硅胶柱。nmr定转化率(>99%),运用高效液相色谱(hplc)测定反应的对映选择性ee值(97%)。
实施例7:
配体6a和[ir(cod)cl]2原位催化轴手性内酯的不对称氢化反应:
在氩气氛围的手套箱内,配体l2(7.9mg,0.0105mmol)和[ir(cod)cl]2(3.4mg,0.005mmol)加入2.5ml小瓶中,用thf(1.0ml)溶解后室温络合1h得催化剂溶液。取催化剂溶液(20ul,0.0002mmol)加入5ml的氢化小瓶中,然后依次加入k2co3(1.6mg),底物内酯(27mg,0.1mmol),0.5mlthf。将氢化小瓶放入氢化反应釜中,氢气置换三次后充入50atmh2,在室温下反应24h。反应完毕释放氢气后,反应混合物过一个短的硅胶柱。nmr定转化率(>99%),运用高效液相色谱(hplc)测定反应的对映选择性ee值(98%)。
参照该方法,多个不对称氢化反应中间体的对映选择性ee值参见图1所示。
以上内容是结合具体的优选实施方式对本发明所作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明。对于本发明所属技术领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单推演或替换,都应当视为属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!