一种用于免疫细胞表面修饰的糖类衍生物及其应用的制作方法
本发明属于化学工程免疫细胞治疗技术领域,具体涉及一种用于免疫细胞表面修饰的糖类衍生物及其应用。
背景技术:
癌症是严重威胁人类健康的杀手,也是目前难以完全攻克的科学难题,一旦患上癌症,几乎等于宣判死亡。传统的治疗方式如手术、放疗和化疗在一定程度上能遏制癌症的发展,延长病人的生存期,但是仍然不能彻底攻克癌症顽疾。进入21世纪以后,随着肿瘤免疫学作为新兴交叉领域的深入发展,肿瘤免疫疗法作为一种崭新的肿瘤治疗方式应运而生。肿瘤免疫疗法掀起了肿瘤治疗的绿色革命,有望彻底攻克肿瘤顽疾,有着广阔的发展前景。
免疫细胞治疗是肿瘤免疫疗法中非常具有潜力的方向之一。主要包括细胞因子激活的杀伤细(lak)、肿瘤浸润淋巴细胞(til)、细胞因子诱导的杀伤细胞(cik)、树突状抗原呈递细胞刺激细胞因子诱导的杀伤细胞(dc-cik)、肿瘤靶向抗体t细胞(tcr-t)、嵌合抗原受体t细胞(car-t)。car-t免疫细胞治疗是当前科技中唯一有可能彻底清除癌细胞的方法,也是目前研究的热点。car-t细胞具有很强的肿瘤杀伤能力,但是由于其过强的免疫杀伤作用,能在短时间内局部消融大量肿瘤细胞,并在局部产生炎症反应,释放出大量的细胞因子,造成细胞因子风暴,严重时甚至会引起脏器功能的衰竭,危害病人的生命。而且car-t需要复杂的基因工程操作,耗时较长,只能用于个体化治疗,无法实现广泛的商业应用,价格相当昂贵。
技术实现要素:
本发明的目的在于克服现有技术缺陷,提供一种用于免疫细胞表面修饰的糖类衍生物。
本发明的另一目的在于提供上述糖类衍生物的应用。
本发明的糖类衍生物基于化学工程免疫细胞治疗技术,与免疫细胞共培养后,能够通过该免疫细胞的代谢完成对该免疫细胞表面的修饰,以使表面修饰后的该免疫细胞能够在体内特异性靶向特定的肿瘤细胞或其他病变细胞并对该肿瘤细胞或其他病变细胞进行杀伤。
本发明的技术方案之一:
一种用于免疫细胞表面修饰的糖类衍生物,其结构式为
r1为h或oh,r2为靶向cd抗原分化簇的苯环类取代基、靶向生物素受体的生物素类取代基、靶向叶酸受体的叶酸类取代基或靶向整合素的rgd类取代基;
r2为oh,r1为靶向cd抗原分化簇的苯环类取代基、靶向生物素受体的生物素类取代基、靶向叶酸受体的叶酸类取代基或靶向整合素的rgd类取代基;
r1为靶向cd抗原分化簇的苯环类取代基、靶向生物素受体的生物素类取代基、靶向叶酸受体的叶酸类取代基或靶向整合素的rgd类取代基,r2为靶向cd抗原分化簇的苯环类取代基、靶向生物素受体的生物素类取代基、靶向叶酸受体的叶酸类取代基或靶向整合素的rgd类取代基。
在本发明的一个优选实施方案中,所述苯环类取代基包括
在本发明的一个优选实施方案中,所述生物素类取代基包括
在本发明的一个优选实施方案中,所述叶酸类取代基包括
在本发明的一个优选实施方案中,所述rgd类取代基包括
进一步优选的,所述免疫细胞包括t细胞、nk细胞、巨噬细胞、dc细胞、b细胞和粒细胞。
本发明的技术方案之二:
一种糖类衍生物在改造免疫细胞中的应用,该糖类衍生物的结构式为
r1为h或oh,r2为靶向cd抗原分化簇的苯环类取代基、靶向生物素受体的生物素类取代基、靶向叶酸受体的叶酸类取代基或靶向整合素的rgd类取代基;
r2为oh,r1为靶向cd抗原分化簇的苯环类取代基、靶向生物素受体的生物素类取代基、靶向叶酸受体的叶酸类取代基或靶向整合素的rgd类取代基;
r1为靶向cd抗原分化簇的苯环类取代基、靶向生物素受体的生物素类取代基、靶向叶酸受体的叶酸类取代基或靶向整合素的rgd类取代基,r2为靶向cd抗原分化簇的苯环类取代基、靶向生物素受体的生物素类取代基、靶向叶酸受体的叶酸类取代基或靶向整合素的rgd类取代基。
在本发明的一个优选实施方案中,所述苯环类取代基包括
在本发明的一个优选实施方案中,所述生物素类取代基包括
在本发明的一个优选实施方案中,所述叶酸类取代基包括
在本发明的一个优选实施方案中,所述rgd类取代基包括
进一步优选的,所述免疫细胞包括t细胞、nk细胞、巨噬细胞、dc细胞、b细胞和粒细胞。
本发明的技术方案之三:
一种免疫细胞的改造方法,将免疫细胞与糖类衍生物在培养基中共培养,使该糖类衍生物通过免疫细胞内正常代谢对免疫细胞的表面进行修饰,该糖类衍生物的结构式为
r1为h或oh,r2为靶向cd抗原分化簇的苯环类取代基、靶向生物素受体的生物素类取代基、靶向叶酸受体的叶酸类取代基或靶向整合素的rgd类取代基;
r2为oh,r1为靶向cd抗原分化簇的苯环类取代基、靶向生物素受体的生物素类取代基、靶向叶酸受体的叶酸类取代基或靶向整合素的rgd类取代基;
r1为靶向cd抗原分化簇的苯环类取代基、靶向生物素受体的生物素类取代基、靶向叶酸受体的叶酸类取代基或靶向整合素的rgd类取代基,r2为靶向cd抗原分化簇的苯环类取代基、靶向生物素受体的生物素类取代基、靶向叶酸受体的叶酸类取代基或靶向整合素的rgd类取代基。
在本发明的一个优选实施方案中,所述苯环类取代基包括
在本发明的一个优选实施方案中,所述生物素类取代基包括
在本发明的一个优选实施方案中,所述叶酸类取代基包括
在本发明的一个优选实施方案中,所述rgd类取代基包括
进一步优选的,所述免疫细胞包括t细胞、nk细胞、巨噬细胞、dc细胞、b细胞和粒细胞。
本发明的技术方案之四:
一种用于免疫细胞表面修饰的糖类衍生物,其结构式为
r3为靶向cd抗原分化簇的苯环类取代基、靶向生物素受体的生物素类取代基、靶向叶酸受体的叶酸类取代基或靶向整合素的rgd类取代基。
在本发明的一个优选实施方案中,所述苯环类取代基包括
在本发明的一个优选实施方案中,所述生物素类取代基包括
在本发明的一个优选实施方案中,所述叶酸类取代基包括
在本发明的一个优选实施方案中,所述rgd类取代基包括
进一步优选的,所述免疫细胞包括t细胞、nk细胞、巨噬细胞、dc细胞、b细胞和粒细胞。
本发明的技术方案之五:
一种糖类衍生物在改造免疫细胞中的应用,该糖类衍生物的结构式为
r3为靶向cd抗原分化簇的苯环类取代基、靶向生物素受体的生物素类取代基、靶向叶酸受体的叶酸类取代基或靶向整合素的rgd类取代基。
在本发明的一个优选实施方案中,所述苯环类取代基包括
在本发明的一个优选实施方案中,所述生物素类取代基包括
在本发明的一个优选实施方案中,所述叶酸类取代基包括
在本发明的一个优选实施方案中,所述rgd类取代基包括
进一步优选的,所述免疫细胞包括t细胞、nk细胞、巨噬细胞、dc细胞、b细胞和粒细胞。
本发明的技术方案之六:
一种免疫细胞的改造方法,将免疫细胞与糖类衍生物在培养基中共培养,使该糖类衍生物通过免疫细胞内正常代谢对免疫细胞的表面进行修饰,该糖类衍生物的结构式为
r3为靶向cd抗原分化簇的苯环类取代基、靶向生物素受体的生物素类取代基、靶向叶酸受体的叶酸类取代基或靶向整合素的rgd类取代基。
在本发明的一个优选实施方案中,所述苯环类取代基包括
在本发明的一个优选实施方案中,所述生物素类取代基包括
在本发明的一个优选实施方案中,所述叶酸类取代基包括
在本发明的一个优选实施方案中,所述rgd类取代基包括
进一步优选的,所述免疫细胞包括t细胞、nk细胞、巨噬细胞、dc细胞、b细胞和粒细胞。
本发明的有益效果是:本发明的糖类衍生物为9位或5位靶向取代基修饰的9-碳单糖唾液酸类衍生物及5位靶向取代基修饰的n-乙酰甘露糖胺类衍生物,实现了对各种免疫细胞(t细胞、nk细胞、巨噬细胞、dc细胞、b细胞、粒细胞)的表面修饰,用于肿瘤或其他疾病治疗,能够获得很好的治疗效果,相对于基因工程免疫细胞治疗中的car-t技术,可以高效实现对免疫细胞的改造,并且可以大规模制造类似货架商品,成本较低,具有巨大的商业应用价值。
附图说明
图1为本发明实施例57中的体外细胞毒性实验结果图之一。
图2为本发明实施例57中的体外细胞毒性实验结果图之二。
图3为本发明实施例58中的体外细胞毒性实验结果图之一。
图4为本发明实施例58的体外细胞毒性实验结果图之二。
图5为本发明实施例59中的bpca-neu5ac修饰的nk-92细胞照片。
图6为本发明实施例59中的体外细胞毒性实验结果图。
图7为本发明实施例59中的nsg小鼠生存曲线图。
图8为本发明实施例60中bpca-neu5ac修饰的cd8t细胞照片。
图9为本发明实施例60中的体外细胞毒性实验结果图。
图10为本发明实施例60中的nsg小鼠生存曲线图。
具体实施方式
以下通过具体实施方式结合附图对本发明的技术方案进行进一步的说明和描述。
本发明的糖类衍生物用于化学工程免疫细胞治疗(cect)的步骤如下:
1)免疫细胞的获取及培养
目标效应细胞,主要包括t细胞、nk细胞、巨噬细胞、dc细胞、b细胞、粒细胞等,以杀伤功能最强的cd8t细胞为例,从供体外周血中进行流式细胞分选获取cd8t细胞,采用特定的培养基对其进行增殖培养获取足够的细胞数。
2)将带靶向基团的目标分子与免疫细胞共培养
在细胞培养基中加入一定浓度的带靶向基团的目标分子,与目标效应细胞共培养一定时间,通过细胞正常代谢对目标效应细胞进行靶向标记。对细胞标记效率进行评估与优化。
3)细胞回输
将经靶向标记的免疫细胞以一定数量和频次通过静脉直接注射进血液中,对相应的肿瘤进行治疗。
系列化合物目标分子的详细描述
这类取代基的功能特点描述:
以上取代基仅为一类功能取代基中的代表结构,其中编号为3、4的取代基代表靶向cd169或cd22或其它cd抗原分化簇的苯环类取代基;编号5的取代基代表靶向生物素受体的生物素类取代基;编号6、7的取代基代表靶向叶酸受体的叶酸类取代基;编号8的取代基代表靶向整合素的rgd类取代基。
根据r基的不同,这些一系列化合物目标分子一共可分为四大类:
第一类,当5位c取代基r2为h取代基时,这类化合物为9位氨基取代的n-乙酰神经氨酸(neu5ac)衍生物。结构如下,其中r1可为编号3-8的任意取代基。
第二类,当5位c取代基r2为oh取代基时,这类化合物为9位氨基取代的n-羟乙酰神经氨酸(neu5gc)衍生物。结构如下,其中r1可为编号3-8的任意取代基。
第三类,当9位c取代基r1位oh取代基时,5位c上r2可为编号3-8的任意取代基。结构如下:
第四类,5位c上r2取代基和9位c上r1取代基可为编号3-8的任意取代基。例如,当5位c上r2取代基为编号3的取代基和9位c上r1取代基为编号4的取代基时,结构如下:
第五类,n-乙酰甘露糖胺的5位c取代基。结构如下,其中r3可为编号3-8的任意取代基。这类化合物经胞内代谢后可转化为第三类化合物。
系列化合物目标分子的胞内代谢原理
这一系列化合物目标分子都可以经胞内唾液酸代谢途径最终呈递到细胞表面,以第五类n-乙酰甘露糖胺的5位c取代基衍生物的代谢途径为例,当r3为h时,此时化合物为乙酰甘露糖胺(mannac),其在细胞质中的乙酰甘露糖胺激酶(mannackinase)催化下可转化为mannac-6-phosphate,然后在neu5ac9-phosphatesynthase催化下与pep(phosphoenolpyruvate)缩合转化为neu5ac-9-phosphate,然后在neu5ac9-phosphatephosphatase催化下脱去一分子磷酸形成neu5ac,然后在细胞核内转化成cmp-neu5ac,转移到高尔基体中添加到糖蛋白或糖脂的末端,然后高尔基体分泌囊泡与细胞膜融合,这样就完成了到细胞膜表面的呈递,成功的对细胞表面进行了修饰。
第一类化合物的合成过程
实施例1
1、将唾液酸(1)(15.0g,50mmol)、三氟乙酸(1ml)与无水甲醇(300ml)混合,将混合物在室温下搅拌至澄清。蒸发除去溶剂,得到唾液酸甲酯,为白色固体,无需进一步纯化。将固体与p2o5在真空干燥过夜,并与无水吡啶共蒸发两次以除去痕量的水,然后溶于无水吡啶(200ml)中。将溶液冷却至0℃,加入对甲苯磺酰氯(10.0g,53mmol)。将混合物加热至室温并搅拌过夜。减压加热除去溶剂,将残余物通过柱层析(20∶1etoac∶meoh)纯化,得到白色固体的化合物(2)(18.0g,38mmol),产率为76%。
2、叠氮化钠(8.0g),化合物(2)(14.0g,30mmol)加入dmf(150ml)中。将溶液加热至80℃过夜。完全除去溶剂,残余物通过柱层析(20∶1etoac∶meoh)纯化,得到化合物(3),为浅黄色固体(8.5g,25mmol,83%)。
3、将化合物(3)(3.0g,8.6mmol)、pd/c(300mg)与无水甲醇(100ml)混合均匀,加入乙酰氯调节至ph≈1-2;将混合物置于h2气氛下搅拌。通过tlc监测至反应完全时(≈12h),将反应物过滤以除去催化剂。蒸发除去溶剂,将残余物、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物6(2.1g,8.6mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(4),为浅黄色固体(2.6g,4.8mmol,55%)。
4、将化合物(4)(548mg,1mmol)、甲醇(5ml)和2mnaoh水溶液(5ml)混合均匀。将溶液在室温下搅拌20分钟,然后用hcl水溶液(2m)调节至ph=7。完全除去溶剂,残余物通过柱层析(2∶1ch2cl2∶meoh)纯化,得到化合物(5),为浅黄色固体(294mg,0.55mmol,55%)。
实施例2
1、将唾液酸(1)(15.0g,50mmol)、三氟乙酸(1ml)与无水甲醇(300ml)混合,将混合物在室温下搅拌至澄清。蒸发除去溶剂,得到唾液酸甲酯,为白色固体,无需进一步纯化。将固体与p2o5在真空干燥过夜,并与无水吡啶共蒸发两次以除去痕量的水,然后溶于无水吡啶(200ml)中。将溶液冷却至0℃,加入对甲苯磺酰氯(10.0g,53mmol)。将混合物加热至室温并搅拌过夜。减压加热除去溶剂,将残余物通过柱层析(20∶1etoac∶meoh)纯化,得到白色固体的化合物(2)(18.0g,38mmol),产率为76%。
2、叠氮化钠(8.0g),化合物(2)(14.0g,30mmol)加入dmf(150ml)中。将溶液加热至80℃过夜。完全除去溶剂,残余物通过柱层析(20∶1etoac∶meoh)纯化,得到化合物(3),为浅黄色固体(8.5g,25mmol,83%)。
3、将化合物(3)(3.0g,8.6mmol)、pd/c(300mg)与无水甲醇(100ml)混合均匀,加入乙酰氯调节至ph≈1-2;将混合物置于h2气氛下搅拌。通过tlc监测至反应完全时(≈12h),将反应物过滤以除去催化剂。蒸发除去溶剂,将残余物、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物6(3.8g,8.6mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(4),为浅黄色固体(3.6g,4.8mmol,55%)。
4、将化合物(4)(745mg,1mmol)、甲醇(5ml)和2mnaoh水溶液(5ml)混合均匀。将溶液在室温下搅拌20分钟,然后用hcl水溶液(2m)调节至ph=7。完全除去溶剂,残余物通过柱层析(2∶1ch2cl2∶meoh)纯化,得到化合物(5),为浅黄色固体(402mg,0.55mmol,55%)。
实施例3
1、将唾液酸(1)(15.0g,50mmol)、三氟乙酸(1ml)与无水甲醇(300ml)混合,将混合物在室温下搅拌至澄清。蒸发除去溶剂,得到唾液酸甲酯,为白色固体,无需进一步纯化。将固体与p2o5在真空干燥过夜,并与无水吡啶共蒸发两次以除去痕量的水,然后溶于无水吡啶(200ml)中。将溶液冷却至0℃,加入对甲苯磺酰氯(10.0g,53mmol)。将混合物加热至室温并搅拌过夜。减压加热除去溶剂,将残余物通过柱层析(20∶1etoac∶meoh)纯化,得到白色固体的化合物(2)(18.0g,38mmol),产率为76%。
2、叠氮化钠(8.0g),化合物(2)(14.0g,30mmol)加入dmf(150m1)中。将溶液加热至80℃过夜。完全除去溶剂,残余物通过柱层析(20∶1etoac∶meoh)纯化,得到化合物(3),为浅黄色固体(8.5g,25mmol,83%)。
3、将化合物(3)(3.0g,8.6mmol)、pd/c(300mg)与无水甲醇(100ml)混合均匀,加入乙酰氯调节至ph≈1-2;将混合物置于h2气氛下搅拌。通过tlc监测至反应完全时(≈12h),将反应物过滤以除去催化剂。蒸发除去溶剂,将残余物、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物6(3.9g,8.6mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(4),为浅黄色固体(3.6g,4.8mmol,55%)。
4、将化合物(4)(758mg,1mmol)、甲醇(5ml)和2mnaoh水溶液(5ml)混合均匀。将溶液在室温下搅拌20分钟,然后用hcl水溶液(2m)调节至ph=7。完全除去溶剂,残余物通过柱层析(2∶1ch2cl2∶meoh)纯化,得到化合物(5),为浅黄色固体(409mg,0.55mmol,55%)。
实施例4
1、将唾液酸(1)(15.0g,50mmol)、三氟乙酸(1ml)与无水甲醇(300ml)混合,将混合物在室温下搅拌至澄清。蒸发除去溶剂,得到唾液酸甲酯,为白色固体,无需进一步纯化。将固体与p2os在真空干燥过夜,并与无水吡啶共蒸发两次以除去痕量的水,然后溶于无水吡啶(200ml)中。将溶液冷却至0℃,加入对甲苯磺酰氯(10.0g,53mmol)。将混合物加热至室温并搅拌过夜。减压加热除去溶剂,将残余物通过柱层析(20∶1etoac∶meoh)纯化,得到白色固体的化合物(2)(18.0g,38mmol),产率为76%。
2、叠氮化钠(8.0g),化合物(2)(14.0g,30mmol)加入dmf(150ml)中。将溶液加热至80℃过夜。完全除去溶剂,残余物通过柱层析(20∶1etoac∶meoh)纯化,得到化合物(3),为浅黄色固体(8.5g,25mmol,83%)。
3、将化合物(3)(3.0g,8.6mmol)、pd/c(300mg)与无水甲醇(100ml)混合均匀,加入乙酰氯调节至ph≈1-2;将混合物置于h2气氛下搅拌。通过tlc监测至反应完全时(≈12h),将反应物过滤以除去催化剂。蒸发除去溶剂,将残余物、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物6(5.5g,8.6mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(4),为浅黄色固体(4.5g,4.8mmol,55%)。
4、将化合物(4)(951mg,1mmol)、甲醇(5ml)和2mnaoh水溶液(5ml)混合均匀。将溶液在室温下搅拌20分钟,然后用hcl水溶液(2m)调节至ph=7。完全除去溶剂,残余物通过柱层析(2∶1ch2cl2∶meoh)纯化,得到化合物(5),为浅黄色固体(515mg,0.55mmol,55%)。
第二类化合物的合成过程
实施例5
1、将唾液酸(1)(15.0g,50mmol)、三氟乙酸(1ml)与无水甲醇(300ml)混合,将混合物在室温下搅拌至澄清。蒸发除去溶剂,得到唾液酸甲酯,为白色固体,无需进一步纯化。将固体与p2o5在真空干燥过夜,并与无水吡啶共蒸发两次以除去痕量的水,然后溶于无水吡啶(200ml)中。将溶液冷却至0℃,加入对甲苯磺酰氯(10.0g,53mmol)。将混合物加热至室温并搅拌过夜。减压加热除去溶剂,将残余物通过柱层析(20∶1etoac∶meoh)纯化,得到白色固体的化合物(2)(18.0g,38mmol),产率为76%。
2、叠氮化钠(8.0g),化合物(2)(14.0g,30mmol)加入dmf(150ml)中。将溶液加热至80℃过夜。完全除去溶剂,残余物通过柱层析(20∶1etoac∶meoh)纯化,得到化合物(3),为浅黄色固体(8.5g,25mmol,83%)。
3、将化合物(3)(3.0g,8.6mmol)、pd/c(300mg)与无水甲醇(100ml)混合均匀,加入乙酰氯调节至ph≈1-2;将混合物置于h2气氛下搅拌。通过tlc监测至反应完全时(≈12h),将反应物过滤以除去催化剂。蒸发除去溶剂,将残余物、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物6(2.1g,8.6mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(4),为浅黄色固体(2.7g,4.8mmol,55%)。
4、将化合物(4)(564mg,1mmol)、甲醇(5ml)和2mnaoh水溶液(5ml)混合均匀。将溶液在室温下搅拌20分钟,然后用hcl水溶液(2m)调节至ph=7。完全除去溶剂,残余物通过柱层析(2∶1ch2cl2∶meoh)纯化,得到化合物(5),为浅黄色固体(302mg,0.55mmol,55%)。
实施例6
1、将唾液酸(1)(15.0g,50mmol)、三氟乙酸(1ml)与无水甲醇(300ml)混合,将混合物在室温下搅拌至澄清。蒸发除去溶剂,得到唾液酸甲酯,为白色固体,无需进一步纯化。将固体与p2o5在真空干燥过夜,并与无水吡啶共蒸发两次以除去痕量的水,然后溶于无水吡啶(200ml)中。将溶液冷却至0℃,加入对甲苯磺酰氯(10.0g,53mmol)。将混合物加热至室温并搅拌过夜。减压加热除去溶剂,将残余物通过柱层析(20∶1etoac∶meoh)纯化,得到白色固体的化合物(2)(18.0g,38mmol),产率为76%。
2、叠氮化钠(8.0g),化合物(2)(14.0g,30mmol)加入dmf(150ml)中。将溶液加热至80℃过夜。完全除去溶剂,残余物通过柱层析(20∶1etoac∶meoh)纯化,得到化合物(3),为浅黄色固体(8.5g,25mmol,83%)。
3、将化合物(3)(3.0g,8.6mmol)、pd/c(300mg)与无水甲醇(100ml)混合均匀,加入乙酰氯调节至ph≈1-2;将混合物置于h2气氛下搅拌。通过tlc监测至反应完全时(≈12h),将反应物过滤以除去催化剂。蒸发除去溶剂,将残余物、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物6(3.7g,8.6mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(4),为浅黄色固体(3.6g,4.8mmol,55%)。
4、将化合物(4)(761mg,1mmol)、甲醇(5ml)和2mnaoh水溶液(5ml)混合均匀。将溶液在室温下搅拌20分钟,然后用hcl水溶液(2m)调节至ph=7。完全除去溶剂,残余物通过柱层析(2∶1ch2cl2∶meoh)纯化,得到化合物(5),为浅黄色固体(410mg,0.55mmol,55%)。
实施例7
1、将唾液酸(1)(15.0g,50mmol)、三氟乙酸(1ml)与无水甲醇(300ml)混合,将混合物在室温下搅拌至澄清。蒸发除去溶剂,得到唾液酸甲酯,为白色固体,无需进一步纯化。将固体与p2os在真空干燥过夜,并与无水吡啶共蒸发两次以除去痕量的水,然后溶于无水吡啶(200ml)中。将溶液冷却至0℃,加入对甲苯磺酰氯(10.0g,53mmol)。将混合物加热至室温并搅拌过夜。减压加热除去溶剂,将残余物通过柱层析(20∶1etoac∶meoh)纯化,得到白色固体的化合物(2)(18.0g,38mmol),产率为76%。
2、叠氮化钠(8.0g),化合物(2)(14.0g,30mmol)加入dmf(150ml)中。将溶液加热至80℃过夜。完全除去溶剂,残余物通过柱层析(20∶1etoac∶meoh)纯化,得到化合物(3),为浅黄色固体(8.5g,25mmol,83%)。
3、将化合物(3)(3.0g,8.6mmol)、pd/c(300mg)与无水甲醇(100ml)混合均匀,加入乙酰氯调节至ph≈1-2;将混合物置于h2气氛下搅拌。通过tlc监测至反应完全时(≈12h),将反应物过滤以除去催化剂。蒸发除去溶剂,将残余物、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物6(3.9g,8.6mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(4),为浅黄色固体(3.7g,4.8mmol,55%)。
4、将化合物(4)(774mg,1mmol)、甲醇(5ml)和2mnaoh水溶液(5ml)混合均匀。将溶液在室温下搅拌20分钟,然后用hcl水溶液(2m)调节至ph=7。完全除去溶剂,残余物通过柱层析(2∶1ch2cl2∶meoh)纯化,得到化合物(5),为浅黄色固体(418mg,0.55mmol,55%)。
实施例8
1、将唾液酸(1)(15.0g,50mmol)、三氟乙酸(1ml)与无水甲醇(300ml)混合,将混合物在室温下搅拌至澄清。蒸发除去溶剂,得到唾液酸甲酯,为白色固体,无需进一步纯化。将固体与p2o5在真空干燥过夜,并与无水吡啶共蒸发两次以除去痕量的水,然后溶于无水吡啶(200ml)中。将溶液冷却至0℃,加入对甲苯磺酰氯(10.0g,53mmol)。将混合物加热至室温并搅拌过夜。减压加热除去溶剂,将残余物通过柱层析(20∶1etoac∶meoh)纯化,得到白色固体的化合物(2)(18.0g,38mmol),产率为76%。
2、叠氮化钠(8.0g),化合物(2)(14.0g,30mmol)加入dmf(150ml)中。将溶液加热至80℃过夜。完全除去溶剂,残余物通过柱层析(20∶1etoac∶meoh)纯化,得到化合物(3),为浅黄色固体(8.5g,25mmol,83%)。
3、将化合物(3)(3.0g,8.6mmol)、pd/c(300mg)与无水甲醇(100ml)混合均匀,加入乙酰氯调节至ph≈1-2;将混合物置于h2气氛下搅拌。通过tlc监测至反应完全时(≈12h),将反应物过滤以除去催化剂。蒸发除去溶剂,将残余物、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物6(5.5g,8.6mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(4),为浅黄色固体(4.6g,4.8mmol,55%)。
4、将化合物(4)(967mg,1mmol)、甲醇(5ml)和2mnaoh水溶液(5ml)混合均匀。将溶液在室温下搅拌20分钟,然后用hcl水溶液(2m)调节至ph=7。完全除去溶剂,残余物通过柱层析(2∶1ch2cl2∶meoh)纯化,得到化合物(5),为浅黄色固体(524mg,0.55mmol,55%)。
第三类化合物的合成过程
实施例9
将化合物(1)(2.67g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(2.5g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(2.8g,5.5mmol,55%)。
实施例10
将化合物(1)(2.67g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(2.9g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(2.9g,5.5mmol,55%)。
实施例11
将化合物(1)(2.67g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(3.01g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(3.02g,5.5mmol,55%)。
实施例12
将化合物(1)(2.67g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(4.98g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(4.11g,5.5mmol,55%)。
实施例13
将化合物(1)(2.67g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(5.11g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(4.18g,5.5mmol,55%)。
实施例14
将化合物(1)(2.67g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(7.04g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(5.24g,5.5mmol,55%)。
第四类化合物的合成过程
实施例15
将化合物(1)(5.03g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(1.98g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(3.75g,5.5mmol,55%)。
实施例16
将化合物(1)(5.03g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(2.32g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(3.94g,5.5mmol,55%)。
实施例17
将化合物(1)(5.03g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(2.44g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(4.01g,5.5mmol,55%)。
实施例18
将化合物(1)(5.03g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(4.41g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(5.10g,5.5mmol,55%)。
实施例19
将化合物(1)(5.03g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(4.54g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(5.16g,5.5mmol,55%)。
实施例20
将化合物(1)(5.03g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(6.47g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(6.23g,5.5mmol,55%)。
实施例21
将化合物(1)(5.37g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(1.98g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(3.94g,5.5mmol,55%)。
实施例22
将化合物(1)(5.37g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(2.32g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(4.13g,5.5mmol,55%)。
实施例23
将化合物(1)(5.37g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(2.44g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(4.20g,5.5mmol,55%)。
实施例24
将化合物(1)(5.37g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(4.41g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(5.28g,5.5mmol,55%)。
实施例25
将化合物(1)(5.37g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(4.54g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(5.35g,5.5mmol,55%)。
实施例26
将化合物(1)(5.37g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(6.47g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(6.41g,5.5mmol,55%)。
实施例27
将化合物(1)(5.49g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(1.98g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(4.01g,5.5mmol,55%)。
实施例28
将化合物(1)(5.49g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(2.32g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(4.19g,5.5mmol,55%)。
实施例29
将化合物(1)(5.49g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(2.44g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(4.26g,5.5mmol,55%)。
实施例30
将化合物(1)(5.49g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(4.41g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(5.35g,5.5mmol,55%)。
实施例31
将化合物(1)(5.49g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(4.54g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(5.42g,5.5mmol,55%)。
实施例32
将化合物(1)(5.49g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(6.47g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(6.48g,5.5mmol,55%)。
实施例33
将化合物(1)(7.47g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(1.98g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(5.01g,5.5mmol,55%)。
实施例34
将化合物(1)(7.47g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(2.32g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(5.29g,5.5mmol,55%)。
实施例35
将化合物(1)(7.47g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(2.44g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(5.35g,5.5mmol,55%)。
实施例36
将化合物(1)(7.47g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(3.14g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(5.73g,5.5mmol,55%)。
实施例37
将化合物(1)(7.47g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(3.26g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(5.80g,5.5mmol,55%)。
实施例38
将化合物(1)(7.47g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(6.47g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(7.57g,5.5mmol,55%)。
实施例39
将化合物(1)(7.60g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(1.98g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(5.16g,5.5mmol,55%)。
实施例40
将化合物(1)(7.60g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(2.32g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(5.36g,5.5mmol,55%)。
实施例41
将化合物(1)(7.60g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(2.44g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(5.42g,5.5mmol,55%)。
实施例42
将化合物(1)(7.60g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(3.14g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(5.80g,5.5mmol,55%)。
实施例43
将化合物(1)(7.60g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(3.26g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(5.87g,5.5mmol,55%)。
实施例44
将化合物(1)(7.60g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(6.47g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(7.64g,5.5mmol,55%)。
实施例45
将化合物(1)(9.52g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(1.98g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(6.23g,5.5mmol,55%)。
实施例46
将化合物(1)(9.52g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(2.32g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(6.41g,5.5mmol,55%)。
实施例47
将化合物(1)(9.52g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(2.44g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(6.48g,5.5mmol,55%)。
实施例48
将化合物(1)(9.52g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(3.14g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(6.86g,5.5mmol,55%)。
实施例49
将化合物(1)(9.52g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(3.26g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(6.92g,5.5mmol,55%)。
实施例50
将化合物(1)(9.52g,10mmol)、edc·hcl(2.0g,10.3mmol)、hobt(1.4g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(6.47g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(10∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(8.69g,5.5mmol,55%)。
第五类化合物的合成过程
实施例51
将化合物(1)(2.15g,10mmol)、edc·hcl(2.0g,10.3mmol)、nhs(1.18g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(2.55g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(7∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(3.83g,9.2mmol,92%)。
实施例52
将化合物(1)(2.15g,10mmol)、edc·hcl(2.0g,10.3mmol)、nhs(1.18g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(2.89g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(7∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(4.14g,9.2mmol,92%)。
实施例53
将化合物(1)(2.15g,10mmol)、edc·hcl(2.0g,10.3mmol)、nhs(1.18g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(3.01g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(7∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(4.25g,9.2mmol,92%)。
实施例54
将化合物(1)(2.15g,10mmol)、edc·hcl(2.0g,10.3mmol)、nhs(1.18g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(4.98g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(7∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(6.06g,9.2mmol,92%)。
实施例55
将化合物(1)(2.15g,10mmol)、edc·hcl(2.0g,10.3mmol)、nhs(1.18g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(5.11g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(7∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(6.18g,9.2mmol,92%)。
实施例56
将化合物(1)(2.15g,10mmol)、edc·hcl(2.0g,10.3mmol)、nhs(1.18g,10.3mmol)、三乙胺(3.6ml,25.8mmol)、化合物(2)(7.04g,10mmol)与dmf(150ml)混合均匀。将混合物在黑暗中搅拌48h。蒸发除去溶剂,残余物通过柱层析(7∶1ch2cl2∶meoh)纯化,得到化合物(3),为浅黄色固体(7.96g,9.2mmol,92%)。
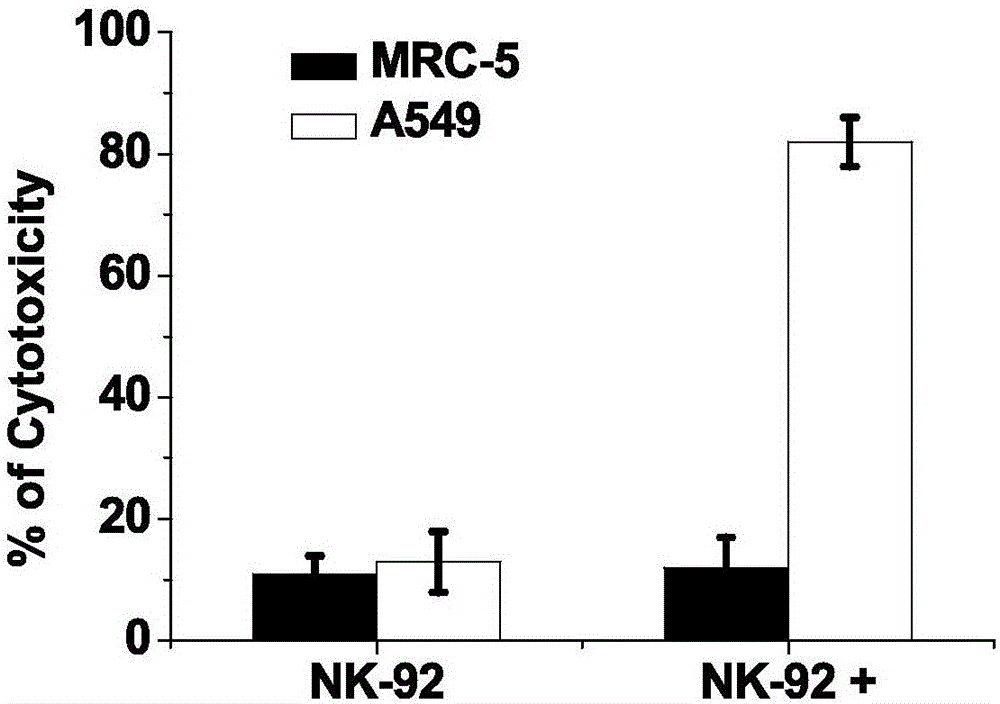
实施例57化合物3.1.1(实施例1制得的化合物(5))修饰的免疫细胞对肺癌(a549)的细胞治疗效果
1、化合物3.1.1修饰nk-92细胞后对肺癌的治疗效果
在α-mem培养基(含12.5%新生牛血清、12.5%马血清、100u/mlrecombinantil-2)中于37℃、5%co2孵育箱中正常培养atcc来源的nk-92(
将mrc-5细胞或者a549细胞与nk-92细胞以数量比1∶10的比例混合,然后在37℃培养箱中共孵育24h后,用乳酸脱氢酶检测试剂盒检测上清液中乳酸脱氢酶的含量,参照空白组计算细胞毒性。结果如图1所示。
乳酸脱氢酶ldh是存在于细胞质的一种酶,当细胞膜受到损伤时,ldh会释放到培养基中,nk细胞与靶细胞结合时,会释放穿孔素及nk细胞毒因子等,破坏靶细胞膜,从而造成ldh的释放,因此检测培养基中ldh的含量即可显示nk细胞的杀伤毒性。从图1可以看出,由于人胚胎肺成纤维细胞(mrc-5)表面生物素受体表达呈阴性,因此原始nk-92及经化合物3.1.1修饰后的nk-92细胞(nk-92+)对其均没有明显的杀伤活性。而人肺癌细胞(a549)高表达生物素受体,经化合物3.1.1修饰后的nk-92细胞(nk-92+)对其杀伤毒性明显增加。
2、化合物3.1.1修饰cd8t细胞后对肺癌的治疗效果
从健康的捐献者外周血中采用流式分选cd8t细胞,采用rpmi-1640培养基(含10%fbs、100iu/mlrecombinantil-2)于37℃、5%co2孵育箱中正常培养,之后更换为含100μm化合物3.1.1的rpmi-1640培养基继续培养72h。更换培养基清洗三次,即得到化合物3.1.1修饰的cd8t细胞(cd8t+)。
将mrc-5细胞或者a549细胞与cd8t细胞以数量比1∶10的比例混合,然后在37℃培养箱中共孵育24h后,用乳酸脱氢酶检测试剂盒检测上清液中乳酸脱氢酶的含量,参照空白组计算细胞毒性。结果如图2所示。
乳酸脱氢酶ldh是存在于细胞质的一种酶,当细胞膜受到损伤时,ldh会释放到培养基中,cd8t细胞与靶细胞结合时,会释放穿孔素及颗粒酶等,破坏靶细胞膜,从而造成ldh的释放,因此检测培养基中ldh的含量即可显示cd8t细胞的杀伤毒性。从图2可以看出,由于人胚胎肺成纤维细胞(mrc-5)表面生物素受体表达呈阴性,因此原始cd8t细胞及经化合物3.1.1修饰的cd8t细胞(cd8t+)对其均没有明显的杀伤活性。而人肺癌细胞(a549)高表达生物素受体,经化合物3.1.1修饰的cd8t细胞(cd8t+)对其杀伤毒性明显增加。
实施例58化合物3.1.2(实施例2制得的化合物(5))修饰的免疫细胞对宫颈癌(hela)的细胞治疗效果
1、化合物3.1.2修饰nk-92细胞后对宫颈癌(hela)的治疗效果
在α-mem培养基(含12.5%新生牛血清、12.5%马血清、100u/mlrecombinantil-2)中于37℃、5%co2孵育箱中正常培养atcc来源的nk-92(
将hela细胞或者a549细胞与nk-92细胞以数量比1∶10的比例混合,然后在37℃培养箱中共孵育24h后,用乳酸脱氢酶检测试剂盒检测上清液中乳酸脱氢酶的含量,参照空白组计算细胞毒性。结果如图3所示。
乳酸脱氢酶ldh是存在于细胞质的一种酶,当细胞膜受到损伤时,ldh会释放到培养基中,nk细胞与靶细胞结合时,会释放穿孔素及nk细胞毒因子等,破坏靶细胞膜,从而造成ldh的释放,因此检测培养基中ldh的含量即可显示nk细胞的杀伤毒性。从图3可以看出,由于人肺癌细胞(a549)表面叶酸受体表达呈阴性,因此原始nk-92及经化合物3.1.2修饰后的nk-92细胞(nk-92+)对其均没有明显的杀伤活性。而人宫颈癌细胞(hela)高表达叶酸受体,经化合物3.1.2修饰后的nk-92细胞(nk-92+)对其杀伤毒性明显增加。
2、化合物3.1.2修饰cd8t细胞后对宫颈癌(hela)的治疗效果
从健康的捐献者外周血中采用流式分选cd8t细胞,采用rpmi-1640培养基(含10%fbs、100iu/mlrecombinantil-2)于37℃、5%co2孵育箱中正常培养,之后更换为含100μm化合物3.1.2的rpmi-1640培养基继续培养72h。更换培养基清洗三次,即得到化合物3.1.2修饰的cd8t细胞(cd8t+)。
将hela细胞或者a549细胞与cd8t细胞以数量比1∶10的比例混合,然后在37℃培养箱中共孵育24h后,用乳酸脱氢酶检测试剂盒检测上清液中乳酸脱氢酶的含量,参照空白组计算细胞毒性。结果如图4所示。
乳酸脱氢酶ldh是存在于细胞质的一种酶,当细胞膜受到损伤时,ldh会释放到培养基中,cd8t细胞与靶细胞结合时,会释放穿孔素及颗粒酶等,破坏靶细胞膜,从而造成ldh的释放,因此检测培养基中ldh的含量即可显示cd8t细胞的杀伤毒性。从图4可以看出,由于人肺癌细胞(a549)表面叶酸受体表达呈阴性,因此原始cd8t细胞及经化合物3.1.2修饰后的cd8t细胞(cd8t+)对其均没有明显的杀伤活性。而人宫颈癌细胞(hela)高表达叶酸受体,经化合物3.1.2修饰后的cd8t细胞(cd8t+)对其杀伤毒性明显增加。
实施例59bpca-neu5ac修饰的nk-92细胞用于b淋巴瘤治疗
1、bpca-neu5ac修饰nk-92细胞
bpca-neu5ac的合成参考synthesisof9-substitutedsialicacidsasprobesforcd22-ligandinteractionsonbcells.shoufahan,briane.collins,andjamesc.paulson.2007。
在α-mem培养基(含12.5%新生牛血清、12.5%马血清、100u/mlrecombinantil-2)中于37℃、5%co2孵育箱中正常培养atcc来源的nk-92(
2、nk-92细胞表面bpca-neu5ac定位分析
用含0.2mg/ml的rhcd22-hisprotein(11958h08h5)的α-mem培养基与上述nk-92细胞于37℃下共孵育2h。更换培养基清洗三次,然后以1∶50的体积比加入6x-histagantibody-dye(ma1-135-a555)对nk-92细胞进行荧光标记,37℃下共孵育1h后进行共聚焦观察(555nm激发,565nm发射)。结果如图5所示。
由于bpca-neu5ac可以有效的靶向cd22,而bpca-neu5ac经胞内唾液酸代谢途径后可以呈递在细胞表面,因此可以与rhcd22-hisprotein有效结合,经荧光探针标记后可以显示其位置,从图5可以看出,nk-92细胞与bpca-neu5ac共孵育后可以有效的呈递到细胞表面,这对于靶向cd22阳性细胞从而发挥杀伤功能非常重要。
3、体外细胞毒性试验
将hela细胞或者rajiburkitt淋巴细胞与nk-92细胞以数量比1∶10的比例混合,然后在37℃培养箱中共孵育24h后,用乳酸脱氢酶检测试剂盒检测上清液中乳酸脱氢酶的含量,参照空白组计算细胞毒性。结果如图6所示。
乳酸脱氢酶ldh是存在于细胞质的一种酶,当细胞膜受到损伤时,ldh会释放到培养基中,nk细胞与靶细胞结合时,会释放穿孔素及nk细胞毒因子等,破坏靶细胞膜,从而造成ldh的释放,因此检测培养基中ldh的含量即可显示nk细胞的杀伤毒性。从图6可以看出,由于hela细胞表面没有cd22受体,因此原始nk-92及经bpca-neu5ac修饰后的nk-92细胞对其均没有明显的杀伤活性。raji是burkitt淋巴瘤细胞,其表面cd22表达呈阳性,经bpca-neu5ac修饰后的nk-92细胞对其杀伤毒性明显增加。
4、对白血病小鼠模型的治疗效果
对6-8周大的nsg小鼠尾静脉注射1×103个raji细胞,构建白血病小鼠模型,然后从第三天开始,每隔两天通过尾静脉注射1×107个nk-92细胞或者经bpca-neu5ac修饰后的nk-92细胞。空白小鼠通过尾静脉注射pbs。每组15只小鼠,观察小鼠存活率。结果如图7所示。
从图7可以看出,白血病小鼠模型建立后小鼠最长存活期为19天左右,尾静脉输入nk-92细胞后可以稍微延长nsg小鼠的存活率,而尾静脉输入经bpca-neu5ac修饰后的nk-92细胞后可将nsg小鼠的最长存活期延长至40天左右。这显示经bpca-neu5ac修饰后的nk-92细胞对体内白血病细胞良好的靶向追踪杀灭效果,其对于进一步的临床试验具有很好的借鉴意义。
实施例60bpca-neu5ac改造的cd8t细胞用于b淋巴瘤治疗
1、bpca-neu5ac修饰cd8t细胞
从健康的捐献者外周血中采用流式分选cd8t细胞,采用rpmi-1640培养基(含10%fbs、100iu/mlrecombinantil-2)于37℃、5%co2孵育箱中正常培养,之后更换为含100μmbpca-neu5ac的rpmi-1640培养基继续培养72h。更换培养基清洗三次,即得到bpca-neu5ac修饰的cd8t细胞。
2、cd8t细胞表面bpca-neu5ac定位分析
用含0.2mg/ml的rhcd22-hisprotein(11958h08h5)的rpmi-1640培养基与上述cd8t细胞于37℃下共孵育2h。更换培养基清洗三次,然后以1∶50的体积比加入6x-histagantibody-dye(ma1-135-a555)对cd8t细胞进行荧光标记,37℃下共孵育1h后进行共聚焦观察(555nm激发,565nm发射)。结果如图8所示。
由于bpca-neu5ac可以有效的靶向cd22,而bpca-neu5ac经胞内唾液酸代谢途径后可以呈递在细胞表面,因此可以与rhcd22-hisprotein有效结合,经荧光探针标记后可以显示其位置,从图8可以看出,cd8t细胞与bpca-neu5ac共孵育后可以有效的呈递到细胞表面,这对于靶向cd22阳性细胞从而发挥杀伤功能非常重要。
3、体外细胞毒性试验
将hela细胞或者rajiburkitt淋巴细胞与cd8t细胞以数量比1∶10的比例混合,然后在37℃培养箱中共孵育24h后,用乳酸脱氢酶检测试剂盒检测上清液中乳酸脱氢酶的含量,参照空白组计算细胞毒性。结果如图9所示。
乳酸脱氢酶ldh是存在于细胞质的一种酶,当细胞膜受到损伤时,ldh会释放到培养基中,cd8t细胞与靶细胞结合时,会释放穿孔素及颗粒酶等,破坏靶细胞膜,从而造成ldh的释放,因此检测培养基中ldh的含量即可显示cd8t细胞的杀伤毒性。从图9可以看出,由于hela细胞表面没有cd22受体,因此原始cd8t细胞及经bpca-neu5ac修饰后的cd8t细胞对其均没有明显的杀伤活性。raji是burkitt淋巴瘤细胞,其表面cd22表达呈阳性,经bpca-neu5ac修饰后的cd8t细胞对其杀伤毒性明显增加。
4、对白血病小鼠模型的治疗效果
对6-8周大的nsg小鼠尾静脉注射1×103个raji细胞,构建白血病小鼠模型,然后从第三天开始,每隔两天通过尾静脉注射1×107个cd8t细胞或者经bpca-neu5ac修饰后的cd8t细胞。空白小鼠通过尾静脉注射pbs。每组15只小鼠,观察小鼠存活率。结果如图10所示。
从图10可以看出,白血病小鼠模型建立后小鼠最长存活期为19天左右,尾静脉输入cd8t细胞后可以稍微延长nsg小鼠的存活率,而尾静脉输入经bpca-neu5ac修饰后的cd8t细胞后可将nsg小鼠的最长存活期延长至42天左右。这显示经bpca-neu5ac修饰后的cd8t细胞对体内白血病细胞良好的靶向追踪杀灭效果,其对于进一步的临床试验具有很好的借鉴意义,而且相对繁琐的car-t治疗具有明显优势。
以上所述,仅为本发明的较佳实施例而已,故不能依此限定本发明实施的范围,即依本发明专利范围及说明书内容所作的等效变化与修饰,皆应仍属本发明涵盖的范围内。
- 还没有人留言评论。精彩留言会获得点赞!