增强细胞的抗癌能力的方法及采用该方法获得的增强型细胞与流程
本发明涉及生物技术领域,更具体而言,本发明涉及一种基于crispr/cas9技术敲除cik细胞的pd-1基因以增强cik细胞的抗肿瘤能力的方法。
背景技术
肝细胞癌(hcc)是世界上癌症相关死亡的最常见原因之一,居所有恶性肿瘤死亡率的第二位,其预后较差。目前传统的肝癌治疗主要涉及部分肝切除、肝移植、射频消融(rfa)和经动脉化疗栓塞。由于大部分病人明确诊断时病情已进入中晚期肝癌阶段,目前传统的治疗方法难以为晚期患者提供临床有效的益处。这也导致了肝癌的5年生存率一直保持在10-20%左右。
为了提高肝癌的存活率,学者们研究出了多种新型治疗方案,其中过继cik细胞治疗是其中有前景的治疗之一。cik细胞是由血液中人外周血单核细胞经体外扩增培养而产生的异质的细胞群,其细胞组成有cd4+t细胞、cd8+t细胞、cd3+cd56+细胞、nk细胞等,其中主要发挥抗肿瘤作用的是cd3+cd56+细胞,其可通过非mhc限制性、nk样细胞杀死机制对肿瘤细胞起到杀伤作用。研究表明cik细胞对多种肿瘤细胞具有杀伤作用,且对正常组织细胞无杀伤功能,这使cik细胞在临床上有广阔的应用前景。目前已有多个cik细胞临床试验正在开展中。例如,lee团队报道cik细胞作为肝癌患者的辅助治疗能明显提高患者的中位生存期,降低肿瘤复发率。
但是,由于cik细胞杀伤功能尚弱,临床上常需要多次输入大量cik细胞或结合其他治疗,这导致病人花费增加。另外有些病人因难以耐受多次输注而中止治疗,导致治疗效果有限。因此近年来,许多研究集中在如何提高cik细胞的能力上。例如,schlimper将cik细胞转染针对cea抗原的特异性嵌合受体来增强抗cea+结肠癌细胞功能。paolaiudicone报道白细胞介素-15可通过先天途径增强cik细胞对上皮癌细胞系的细胞毒性。而通过基因编辑手段给予cik细胞进行相关基因敲除从而增强cik细胞的抗肿瘤能力也逐渐受到学者重视。
pd-1,又名程序死亡因子1,是t细胞表面抑制性受体之一,主要表达于活化的t细胞,同时其也是t细胞耗竭的分子标志之一,在癌症和慢性感染中,其表达上调。越来越多的证据表明,pd-1在体内和体外对cd8+t细胞的效应功能表现出负面影响。先前的研究已经报道了用抗体阻断pd-1增加了肿瘤中的效应t细胞功能并且提高了抗肿瘤效果。
最近聚焦的定期间隔短回文重复序列(crispr)和crispr相关蛋白(cas)(crispr-cas9)系统成为用于基因编辑的有力工具。先前的研究表明,使用cas9rnp(cas9:sgrnaribonucleoprotein)靶向单个基因显著提高了cas9介导的基因组靶向效率并降低了位点依赖性脱靶效应。然而对于非均质的cik细胞,目前尚未见有文献研究敲除pd-1后的cik细胞的功能情况。
技术实现要素:
本发明要解决的问题是,通过cas9rnp介导的敲除细胞毒t淋巴细胞或cik细胞的pd-1基因能否增强细胞的抗肿瘤、特别是抗癌能力。本发明的发明人证实,通过电穿孔cas9rnp进行的pd-1基因敲除在技术上是可行和高效的;此外,细胞毒t淋巴细胞或cik细胞中的pd-1敲除增强了细胞对肝癌细胞系的细胞免疫应答和细胞毒性。
因此,本发明的一个目的是提供基于crispr/cas9技术敲除细胞的pd-1基因以增强细胞的抗癌能力的方法,该方法中采用的靶向pd-1基因的sgrna,以及获得的增强型细胞。
本发明的其他目的是提供sgrna或增强型细胞在制备用于治疗癌症的药物中的用途,以及包含本发明的sgrna的试剂盒。
本发明的技术方案如下。
一方面,本发明提供一种增强细胞的抗癌能力的方法,所述方法包括,基于crispr/cas9技术敲除所述细胞的pd-1基因。
具体而言,本发明的方法包括以下步骤:将cas9蛋白和靶向pd-1基因外显子-1的sgrna电转到细胞中,其中所述细胞为针对肿瘤特异性抗原或肿瘤相关抗原的细胞毒t淋巴细胞或cik细胞。
其中所述sgrna的核苷酸序列如seqidno.1、seqidno.2或seqidno.3所示:
seqidno.1(pd-1sgrna1):gtctgggcggtgctacaact
seqidno.2(pd-1sgrna2):tgtagcaccgcccagacgac
seqidno.3(pd-1sgrna3):accgcccagacgactggcca
优选地,所述sgrna的核苷酸序列如seqidno.1所示。
优选地,所述cas9蛋白和sgrna以1∶0.5-2的质量比进行电转;
优选地,所述cas9蛋白、sgrna和细胞以9.44(μg):9-12(μg):1-5×106个细胞进行电转。
根据本发明的具体实施方式,cas9蛋白使用neb公司的engencas9nls(20um),用量可以为3-5μl(9.44μg-16.1μg);sgrna为体外转录产生的sgrna,用量可以为3-5μl(9μg-15μg)。
根据本发明的具体实施方式,所述cas9蛋白、sgrna和细胞以9.44(μg):9-12(μg):5×106个细胞进行电转。
根据本发明的具体实施方式,采用lonza4d-nucleofectorxunit电转仪以eo-115电转程序进行电转。
优选地,所述细胞为cik细胞,并且其中cd3+cd56+细胞比例≥20%,cd3+cd8+细胞比例≥50%。
根据本发明的具体实施方式,所述方法包括以下步骤:
1)获得cik细胞,其中所述cik细胞的cd3+cd56+细胞比例≥20%,cd3+cd8+细胞比例≥50%;
根据本发明的具体实施方式,可以由外周血中的单个核细胞制备cik细胞;
2)将cas9蛋白和靶向pd-1基因外显子-1的sgrna电转到cik细胞中;其中,所述cas9蛋白、sgrna和cik细胞以9.44(μg):9-12(μg):5×106个细胞并且采用lonza4d-nucleofectorxunit电转仪以eo-115电转程序进行电转;
根据本发明的具体实施方式,电转前,用电转液重悬cik细胞,同时将cas9蛋白和sgrna加入另外的电转液中室温孵育10分钟,然后将cas9蛋白和sgrna混合液加入细胞悬液中,进行电转。
优选地,所述方法还包括以下步骤:
3)将经电转的cik细胞于37℃、5%co2下静置24小时。
根据本发明的具体实施方式,静置后,可以将经电转的cik细胞悬浮于预热的t细胞培养基中,在37℃、5%co2下培养,其中细胞培养基每2天用新鲜的含5%人ab型血浆和100u/ml重组人il-2完全培养基半量换液。
本发明所述的抗癌能力优选为抗肝癌、结直肠癌、乳腺癌、淋巴瘤、白血病、肺癌、胃癌、胰腺癌、后腹膜肿瘤、食管癌、胆管癌、胆囊癌、十二指肠癌、甲状腺癌、鼻咽癌、喉癌、恶性黑色素瘤、子宫内膜癌、宫颈癌、卵巢癌、肾癌、膀胱癌、前列腺癌、骨肉瘤、骨转移癌或恶性胶质瘤等癌症的能力,更优选为抗肝癌能力。
另一方面,本发明提供采用上述方法制得的增强型细胞。其中,优选地,在本发明的增强型细胞中,pd-1基因的敲除率≥40%。优选地,本发明提供采用上述方法制得的增强型cik细胞。
又一方面,本发明还提供用于基于crispr/cas9技术敲除pd-1基因的sgrna,所述sgrna的核苷酸序列如seqidno.1、seqidno.2或seqidno.3所示。优选地,所述sgrna的核苷酸序列如seqidno.1所示。
还一方面,本发明提供一种药物组合物,所述药物组合物包含上述sgrna或增强型细胞。优选地,所述药物组合物包含上述sgrna或增强型cik细胞。
相应地,本发明还提供上述sgrna或增强型细胞在制备用于治疗癌症的药物中的用途;
优选地,所述癌症为肝癌、结直肠癌、乳腺癌、淋巴瘤、白血病、肺癌、胃癌、胰腺癌、后腹膜肿瘤、食管癌、胆管癌、胆囊癌、十二指肠癌、甲状腺癌、鼻咽癌、喉癌、恶性黑色素瘤、子宫内膜癌、宫颈癌、卵巢癌、肾癌、膀胱癌、前列腺癌、骨肉瘤、骨转移癌或恶性胶质瘤等,优选为肝癌。
另一方面,本发明提供一种用于基于crispr/cas9技术敲除细胞、优选cik细胞的pd-1基因的试剂盒,所述试剂盒包括:靶向pd-1基因外显子-1的sgrna;
优选地,所述sgrna的核苷酸序列如seqidno.1、seqidno.2或seqidno.3所示;更优选地,所述sgrna的核苷酸序列如seqidno.1所示。
优选地,所述试剂盒还包括cas9蛋白;
优选地,所述试剂盒还包括用于电转sgrna和/或cas9蛋白的试剂。
本发明提供了基于电穿孔cas9rnp进行的针对肿瘤特异性抗原或肿瘤相关抗原的细胞毒t淋巴细胞或cik细胞的pd-1基因敲除方法,其中采用了筛选出的针对pd-1第1外显子序列的3个sgrna以及特定的电转程序。实验证明,本发明的方法能够使cik细胞的pd-1基因敲除率达到41.23±0.52%,敲除前和敲除后cik细胞的pd-1表达率分别为4.54±0.28%和1.81±0.31%;同时elispot和细胞毒实验显示,本发明提供的敲除pd-1后的cik细胞的ifn-γ分泌功能及对肝癌细胞系的杀伤作用均得到增强。因此,敲除cik细胞的pd-1基因能有效增强cik细胞抗肝癌细胞能力,所制得的增强型cik细胞是肿瘤细胞免疫治疗的一种新的效应细胞。
附图说明
以下,结合附图来详细说明本发明的实施方案,其中:
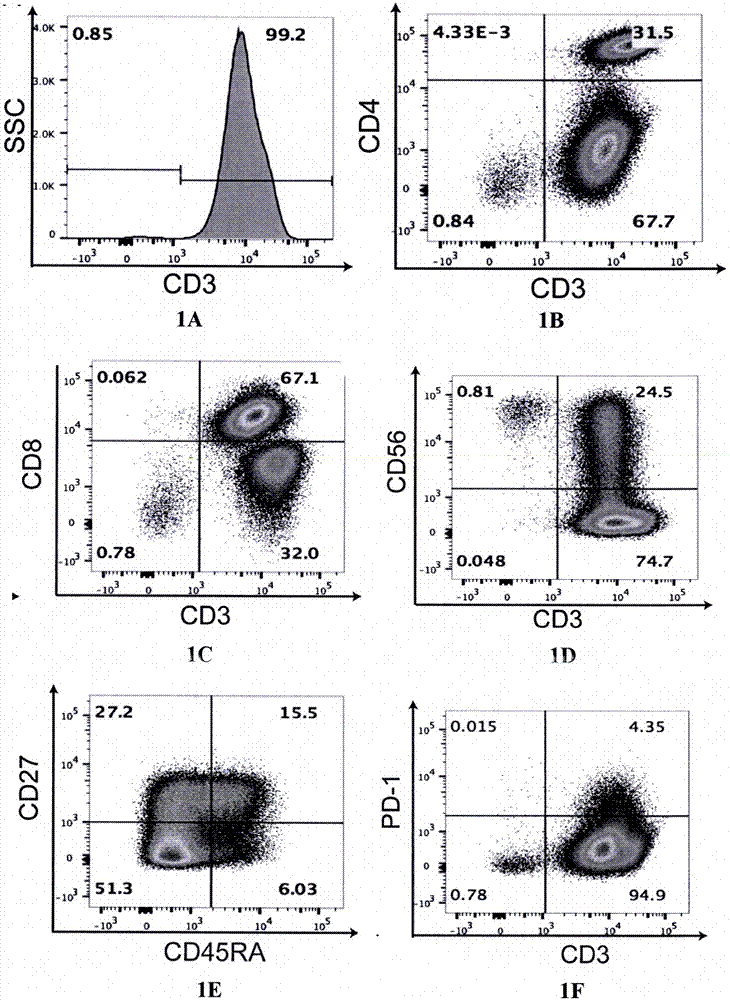
图1显示了体外培养cik细胞的免疫表型的流式细胞术分析。其中,图1a至图1d显示cik细胞标志物cd3、cd4、cd8、cd56的分析结果;图1e显示将cd3+细胞进一步门控cd45ra和cd27,以显示cik细胞的记忆细胞和效应细胞的比例;图1f显示cik细胞中pd-1的表达情况。
图2显示了cik细胞的pd-1敲除情况的检测结果。其中,图2a显示pd-1基因上3个sgrna靶向位点,其中粗斜体表示sgrna导向序列,粗体表示pam序列;图2b显示使用tide分析不同sgrna的pd-1敲除效率(平均值±sem,n=3),实验在三个生物学重复中进行;图2c显示使用t7e1错配酶切分析法检测不同sgrna的pd-1敲除效率,m,marker;wt,野生型;ctrl为对照组,转染无关sgrna+cas9蛋白;图2d显示与野生型序列(wt)相比,pd-1敲除位点的indel情况,其中sgrna靶向位点以粗斜体显示,pam序列以粗体显示,缺失突变以中横线显示,插入突变以下划线显示,将每个样品的pcr产物亚克隆,并对每个克隆的等位基因进行测序。16/40表示在测序的总克隆中含有突变等位基因的克隆的数目。
图3显示了pd-1敲除的cik细胞与野生型cik细胞的免疫表型变化。其中图3a-1和图3a-2显示通过门控cd3+细胞分析pd-1+细胞的结果;图图3b-1和图3b-2显示通过门控cd3+细胞分析cd4+和cd8+细胞的结果;图3c-1和图3c-2显示通过门控cd3+细胞分析cd45ra+/cd27+、cd45ra-/cd27+、cd45ra-/cd27-和cd45ra+/cd27-细胞结果。
图4显示了pd-1敲除的cik细胞和野生型cik细胞的ifn-γ产生测定结果。
图5显示了pd-1敲除的cik细胞和野生型cik细胞对肝癌smmc-7721细胞系的细胞毒性测定结果。
具体实施方式
以下参照具体的实施例来说明本发明。本领域技术人员能够理解,这些实施例仅用于说明发明,其不以任何方式限制本发明的范围。
下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的药材原料、试剂材料等,如无特殊说明,均为市售购买产品。其中,人肝癌细胞系smmc-7721细胞系购买自atcc,在含有10%fbs和100u/ml青霉素、100mg/ml链霉素的dmem高糖培养基中培养。所有细胞均培养于37℃、5%co2细胞培养箱。cas9蛋白为购自neb公司的engencas9nls(20um),可以使用3-5μl(9.44μg-16.1μg)。使用lonza4d-nucleofectorxunit电转仪。
关于统计分析:所有实验独立重复三次。结果以平均值±标准误表示。使用双尾独立t检验来确定统计学意义。使用graphpadprism6软件来评估组间差异的显着性。p值<0.05被认为具有统计学意义。
实施例1cik细胞培养
人外周血取自北京世纪坛医院的肝细胞肝癌患者,已获得肝癌患者的书面同意,并获得医院伦理委员会批准。
cik细胞的培养方法依据前人研究的方案(schmidt-wolfig等人,useofascidmouse/humanlymphomamodeltoevaluatecytokine-inducedkillercellswithpotentantitumorcellactivity.j.exp.med.1991;174(1):139-149)。简而言之,第0天从肝细胞肝癌患者获得40ml外周血,用ficoll-hypaque梯度离心法分离pbmc细胞,将pbmc细胞悬浮于gt-t551无血清培养基(takara公司,日本)中,加入5%人ab型血浆、1000u/mlifn-γ(peprotech)。第1天,加入50ng/ml抗cd3抗体(ebiosciences)、100u/ml重组人il-2(ebioscience)至细胞培养液中。此后每隔2天予以含5%人ab型血浆和100u/ml重组人il-2(e-bioscience)的新鲜gt-t551无血清培养基(takara公司,日本)半量换液,保持细胞浓度为2×106/ml,第15天收集cik细胞,分析cik细胞的表型和细胞毒性,所有产物不含细菌、支原体或真菌,内毒素<5eu。
流式细胞术用于监测体外培养cik细胞的免疫表型。结果如图1所示,cd3+细胞占cik细胞的98.53±1.08%,cd4+细胞占cd3+cik细胞的23±5.541%,cd8+细胞占cd3+cik细胞的58.8±4.834%,和cd56+占cd3+cik细胞的比例达到23.73±2.04%。另外,pd-1+占cd3+cik细胞的比例达到4.54±0.2845%,这与相关文献cik细胞免疫表型大致相仿(oliosop等人,immunotherapywithcytokineinducedkillercellsinsolidandhematopoietictumours:apilotclinicaltrial.hematol.oncol.2009;27(3):130-139),表明成功激活扩增培养cik细胞。
实施例2sgrna设计和sgrna的体外t7转录
从ncbi获得了pd-1第1外显子序列,使用两个crispr设计工具(http://crispr.mit.edu和https://portals.broadinstitute.org/gpp/public/)来设计sgrna,综合挑选出3个grna:
seqidno.1(pd-1sgrna1):gtctgggcggtgctacaact
seqidno.2(pd-1sgrna2):tgtagcaccgcccagacgac
seqidno.3(pd-1sgrna3):accgcccagacgactggcca
使用px330质粒(addgene质粒#4223)作为模板,以t7启动子+20bp靶向序列的寡核苷酸+20bpsgrnascaffer作为正向引物(taatacgactcactatagnnnnnnnnnnnnnnnnnnn(20bp靶序列)gttttagagctagaaatagc)。具体地,三个正向引物分别为:
seqidno.4(pd-1sgrna1的正向引物):
seqidno.5(pd-1sgrna2的正向引物):
seqidno.6(pd-1sgrna3的正向引物):
反向引物为;
seqidno.7(反向引物):agcaccgactcggtgccact。
通过pcr扩增t7-sgrna体外转录模板,吸附柱纯化pcr产物,并使用hiscribet7quickhighyieldrnasynthesiskit(neb,usa)体外转录出sgrna。使用rnaclean&concentratortm-25(zymoresearch,usa)纯化rna并在无rna酶的水中洗脱,洗脱后立即使用或-80℃保存,浓度为3μg/μl。
实施例3制备pd-1敲除的cik细胞并检测敲除情况
使用p3primarycell4d-nucleofectorxkit(lonza,germany)通过4d-nucleofectorsystemx(lonza,germany),用9.44μgcas9蛋白(neb,usa)以及12μg的实施例2制备的三种靶向pd-1外显子1的sgrna中任一种,电穿孔5×106个实施例1中制备的cik细胞。其中电转前预热50ml无血清gt-t551培养基,收集cik细胞,离心并加入70μl电转液重悬细胞,同时将cas9蛋白和sgrna加入30μl电转液中,室温孵育10分钟后,将cas9蛋白和sgrna混合液加入细胞悬液中,使用lonza4d-nucleofectorxunit电转仪以eo-115电转程序进行电转。电穿孔后,细胞静置24小时,然后将细胞重悬于2ml预温的含5%人ab型血浆和100u/ml重组人il-2的gt-t551培养基中,并转移至12孔细胞板中,并在37℃、5%co2培养箱中培养,细胞培养基每2天用新鲜的含5%人ab型血浆和100u/ml重组人il-2完全培养基半量换液。
通过t7el错配修复分析法、tide(通过分解追踪插入)分析和克隆序列分析来检查突变。电转染后第7天收获细胞,用基因组dna提取试剂盒提取基因组dna,使用高保真q5pcr试剂盒扩增pd-1基因敲除位点附近dna片段:
seqidno.8(正向引物pd-f):ccagcactgcctctgtcactctcg;
seqidno.9(反向引物pd-r):acgtcgtaaagccaaggttagtccc。
使用engen突变检测试剂盒(neb,美国)对纯化后的pcr产物进行t7e1错配酶切分析法分析dna突变情况。使用引物pd-f作为测序引物对pcr产物进行sanger测序,测序结果使用网络工具(http://tide.nki.nl)进行tide分析测序。使用pgem-teasyvectorsystems试剂盒(promega,美国)将纯化的pcr片段克隆到pgem-teasy载体中以检测突变体等位基因。每个转化的pcr连接产物样品总共40个菌落使用通用引物m13f进行测序。所用的所有pcr方法均遵循制造商提供的说明书或标准分子克隆方案。
结果参见图2。结果表明,sgrna1的敲除效率最高,达到41.23±0.5239%(tide分析法)。进一步扩增并亚克隆了sgrna1目标区域并鉴定了突变等位基因。发现40个测序等位基因中的16个是突变体,证实了pd-1敲除位点的突变情况,如图2d所示,所有的突变精确地发生在sgrna1靶向区域。这说明成功敲除cik细胞中的pd-1基因。
所获得的pd-1经敲除的cik细胞简称为“pd-1ko/cik细胞”、“pd-1ko-cik细胞”或“增强型cik细胞”。
实施例4增强型cik细胞的性能检测
(一)免疫分型
将5×106个野生型cik细胞及电转后第7天的cik细胞重悬于100μlpbs中,然后加入单克隆抗体cd3-percp-cy5.5、cd4-fitc、cd8-apc、cd56-pe、cd45ro-pe、cd45ra-fitc、cd27-brilliantviolet421、cd279(pd-1)-pe,4℃避光孵育30分钟后,使用facsariar流式细胞仪检测样品。所有抗体均购自bdbioscience(sandiego,ca,usa)。使用flowjov.10.2(treestar,ashland,or,usa)分析流式细胞数据。
使用流式细胞仪检测cd4、cd8、cd45ra、cd27、cd279来评估pd-1敲除后的cik细胞的免疫表型,尽管pd-1的基线表达相对较低,pd-1+cd3+细胞在野生型cik细胞中的百分比为4.54±0.2845%,但是在pd-1基因敲除的cik细胞上为1.81±0.3121%(图3a-1和图3a-2),与野生型cik细胞对比,有明显差异,具有统计学意义。通过cd4和cd8的表达以及初始型t细胞(cd45ra+/cd27+,tn)、中心记忆型t细胞(cd45ra-/cd27+,tcm)、效应记忆型t细胞(cd45ra-/cd27-,tem)和效应t细胞(cd45ra+/cd27-,teff)的特征来评估cik敲除pd-1后的免疫表型(图3b-1和图3b-2,以及图3c-1和图3c-2)。与对照cik细胞相比,在pd-1敲除t细胞中免疫表型大致相同,无明显变化,这说明敲除cik细胞的pd-1不影响cik细胞的正常表型改变。
(二)细胞因子分泌
以e∶t=20∶1比例将5×103个smmc-7721细胞(靶细胞)和1×105个野生型cik细胞或pd-1ko-cik细胞(效应细胞)加入到完全gt-t551培养基中共培养24小时后,弃上清液,使用ifn-γelispot试剂盒(dakewei,中国)通过计算斑点数来比较细胞分泌ifn-γ的能力强弱情况。用elispotctl阅读器(celltechnologyinc,columbia,md)扫描平板,并用elispot软件(aid,strassberg,germany)分析结果。结果如图4所示,pd-1ko-cik细胞的斑点数明显高于野生型cik细胞(90.00±1.528vs24.00±2.517),具有统计学意义。这也表明pd-1的敲除增强了cik细胞对肝癌细胞系的细胞免疫应答。
(三)对肝癌细胞的细胞毒作用
通过荧光素酶介导的细胞毒性分析来检测cik细胞和pd-1ko-cik细胞的细胞毒性。通过慢病毒感染构建带有荧光素酶和gfp蛋白的肝癌smmc-7721细胞(参见王晓敏等人,gfp和luc双标技术在小鼠肿瘤模型建立中的应用,实验动物与比较医学,2010,30(1):2-7),即fluc-egfp/smmc772肝癌细胞,然后将该细胞与效应细胞(野生型cik细胞或pd-1ko/cik细胞)按照不同效靶比混合接种于白色底透的荧光检测96孔板中,其中fluc-egfp/smmc7721肝癌细胞数量固定为5000个细胞,效应细胞按照效靶比10∶1、5∶1、2.5∶1分别加入5×104、2.5×104、1.25×104细胞,于37℃共培养16-18h后,加入100μl底物混合物后立即于多功能酶标仪检测相对荧光强度。
根据下列公式计算细胞杀伤率:(%杀伤率=100-(靶效细胞共培养孔的荧光数值)/(靶细胞孔荧光数值)×100)
结果如图5所示。在效靶比为10∶1、5∶1、2.5∶1时,pd-1ko-cik细胞和野生型cik细胞对肝癌细胞的杀伤率对比分别为90.77±0.72%vs50.23±2.66%,67.03±3.22%vs32.43±1.33%,37.67±4.14%vs22.07±2.63%,p<0.05,差异具有统计学意义。这表明与野生型cik细胞对比,肝癌患者来源的pd-1ko-cik细胞对肝癌smmc-7721的杀伤率更高。这也说明pd-1敲除能增强cik细胞抗肝癌细胞能力。
以上对本发明具体实施方式的描述并不限制本发明,本领域技术人员可以根据本发明作出各种改变或变形,只要不脱离本发明的精神,均应属于本发明所附权利要求的范围。
- 还没有人留言评论。精彩留言会获得点赞!