伪狂犬病病毒基因组Fosmid文库、构建方法及其在构建突变体中的应用与流程
本发明涉及fosmid文库及其构建方法,尤其涉及伪狂犬病病毒基因组fosmid文库及其构建方法,本发明进一步涉及该伪狂犬病病毒基因组fosmid文库在快速构建伪狂犬病病毒突变体中的应用,属于伪狂犬病病毒基因组fosmid文库及其构建和应用领域。
背景技术
伪狂犬病(pseudorabies,pr)是由伪狂犬病病毒(pseudorabiesvirus,prv)引起的猪、羊、牛等多种家畜及野生动物的一种以发热、奇痒(猪除外)、脑脊髓炎、呼吸和神经系统障碍为主要特征的急性传染病(mettenleiter,1989)。猪是本病的贮存宿主和主要传染源,可通过消化道和呼吸道水平传播,也可通过胎盘垂直传播(schoenbaumetal.,1990)。各种日龄的猪均可感染,妊娠母猪感染后常发生流产、死胎、木乃伊胎,其中以产死胎为主要特征;哺乳仔猪感染后出现神经症状、麻痹、衰竭死亡,死亡率几乎高达100%;育肥猪感染后出现呼吸道症状、生长发育不良;成年猪感染耐过后多长期带毒和排毒,成为最危险的传染源,给伪狂犬病的净化带来了极大的困难(antq,pengjm,tianzj,zhaohy,lin,liuym,chenjz,lengcl,suny,changd,tonggz.pseudorabiesvirusvariantinbartha-k61-vaccinatedpigs,china.emerginfectdis.2013,19:1749-755;luoy,lin,congx,wangch,dum,lil,zhaob,yuanj,liudd,lis,liy,suny,qiuhj.pathogenicityandgenomiccharacterizationofapseudorabiesvirusvariantisolatedfrombartha-k61-vaccinatedswinepopulationinchina.vetmicrobiol.2014,174:107-115;suny,luoy,wangch,yuanj,lin,songk,qiuhj.controlofswinepseudorabiesinchina:opportunitiesandlimitations.vetmicrobiol.2016,183:119-124.)。
prv属于疱疹病毒科α疱疹病毒亚科水痘病毒属成员(kingetal.,2012),基因组为线性双链dna,长度约为143kb,g+c含量约为73%。由长独特区(ul)和短独特区(us)以及us两侧的末端反向重复序列(tr)与内部重复序列(ir)组成(hooftvaniddekingeetal.,1996)。prv基因组中含有至少70个基因,可编码70~100种蛋白质,包括结构蛋白、毒力蛋白、复制酶等,这些蛋白作为病毒与细胞相互作用的重要因子,在病毒的吸附、入侵、复制、组装和释放等不同阶段以及在病毒潜伏感染过程中行使不同的功能,一直是prv研究的重点(pomeranzetal.,2005)。但是prv基因组较大,对其基因进行操作以及改造和修饰比较困难。
稳定的大片段基因组文库是生物基因组学研究的材料平台。基因文库是将某一生物的全基因或某部分基因以大小不同的片段克隆到不同的载体,并转移到宿主中。当需要某一基因片段时,可通过某种筛选方法获得包含所需的基因片段的克隆,并可通过宿主的增殖大量获得所需的基因。按载体可插入片段的大小,可将基因文库分为质粒文库、噬菌体文库、fosmid文库、bac文库和yac文库等(kimcg,fujiyamaa,saitoun.constructionofagorillafosmidlibraryanditspcrscreeningsystem.genomics.2003,82:571-575;magriniv,warrenwc,wallisj,goldmanwe,xuj,mardiser,mcphersonjd.fosmid-basedphysicalmappingofthehistoplasmacapsulatumgenome.genomeres.2004,14:1603-1609;moonda,magorke.constructionandcharacterizationofafosmidlibraryforcomparativeanalysisoftheduckgenome.animgenet.2004,35:417-418;李昂,张安,唐君,周志林,赵东兰,曹清河.fosmid基因组文库构建及应用现状.江苏农业科学,2013,41(6):28-30.)。
fosmid文库是应用较广的大片段基因组文库,具有稳定性高、随机性好、拷贝数可控等特点,主要用于基因的克隆、基因组序列测定、物理图谱构建以及比较基因组研究(cunninghamc,hikimaj,jennymj,chapmanrw,fanggc,saskic,lundqvistml,wingra,cupitpm,grossps,warrgw,tomkinsjp.newresourcesformarinegenomics:bacterialartificialchromosomelibrariesfortheeasternandpacificoysters(crassostreavirginicaandc.gigas).marbiotechnol(ny).2006,8:521-533.)。fosmid基因组文库是应用fosmid克隆载体构建而成的文库。fosmid克隆载体是kim等将pbac引入puccos而构建的载体系统(birrenbw,tachi-iriy,kimuj,nguyenm,shizuyah,korenbergj,simon,mi.ahumanchromosome22fosmidresource:mappingandanalysisof96clones.genomics.1996,34:97-106.)。fosmid以大肠杆菌f-因子为基础,以氯霉素抗性基因为选择标记,同时含有λ噬菌体dna的cos序列,可以包装外源dna片段。插入外源片段的粘粒一般为30~40kb。尽管fosmid文库插入的片段小,但是fosmid文库具有稳定性好、没有偏向性、构建周期短以及操作简便等优点,为快速构建突变病毒提供了良好的平台(detomasoaw,weissmanil.constructionandcharacterizationoflarge-insertgenomiclibraries(bacandfosmid)fromtheascidianbotryllusschlosseriandinitialphysicalmappingofahistocompatibilitylocus.marbiotechnol(ny).2003,5:103-115.)。
prv属于疱疹病毒,基因组中包含大量的重复序列,且基因组以不同形式的异构体存在。因而在分析这些重复序列和异构体时,需要对完整的prv基因片段进行分析。然而,prv基因组高达143kb,而且极易断裂,基因组一旦断裂,便失去感染性,无法获得重组病毒。此外,prv的全基因组与bac质粒进行连接,在电转化的过程中往往会出现片段缺失,影响获得重组病毒的效率(lermal,
基于构建的fosmid文库,利用反向遗传操作技术便于在细菌中对庞大的prv基因组进行基因的缺失、插入或突变,能够极大提升获得prv突变体的效率。
因此,提供一种伪狂犬病病毒基因组fosmid文库的快速构建方法,对prv基因进行快速准确的修饰、研究其功能以及开发新型prv重组疫苗等方面均具有重要的意义。
技术实现要素:
本发明目的之一是提供一种伪狂犬病病毒基因组fosmid文库的快速构建方法以及由该构建方法所得到的伪狂犬病病毒基因组fosmid文库;
本发明的目的之二是所构建的伪狂犬病病毒基因组fosmid文库应用于对prv基因进行快速准确的修饰、研究其功能以及开发新型prv重组疫苗等。
本发明的上述目的是通过以下技术方案来实现的:
本发明首先提供了一种伪狂犬病病毒基因组fosmid文库的构建方法,包括:(1)提取伪狂犬病病毒基因组dna;(2)将伪狂犬病病毒基因组dna片段末端补平修饰后与pcc1fos载体连接得到重组dna;(3)将重组dna包装后感染大肠杆菌获得粘粒;(4)筛选用于拯救病毒的粘粒组合,获得伪狂犬病病毒全长基因组的fosmid文库。
优选的,步骤(2)中将所提取的伪狂犬病病毒基因组dna通过物理剪切方式获得随机断裂的基因组dna片段。本发明将prv-tj株基因组dna用物理方法打碎,将巨大的基因组随机片段化再与pcc1fos载体连接得以成功构建prv的fosmid文库。采用该方法构建prv感染性克隆仅需30天左右,而传统的基于bac文库构建prv感染性克隆需要3~4个月,因此,本发明方法显著提高了获得prv感染性克隆的效率。
优选的,步骤(3)中用噬菌体包装蛋白包装重组dna。
优选的,步骤(3)中所述的大肠杆菌为epi300宿主细胞。
根据基因组结构的不同,疱疹病毒科成员被分为a、b、c、d、e和f六种类型。α疱疹病毒根据基因组结构被归为d型或e型。prv基因组ul区两侧缺少重复序列,具有典型的e型基因组特征。在成熟的病毒粒子中疱疹病毒基因组以几种不同的异构体形式存在,具有e型基因组结构的疱疹病毒的ul区颠倒频率较低,故主要以p型、is型两种异构体形式存在(yasuhikoh,richardw.isomerizationoftheulregionofvaricella-zostervirusdna.virusres,1987,8:825-831.)。基因组的异构体化是在病毒dna复制与包装时发生的事件,对病毒的生长无显著影响。同源重组在异构体的产生中起到了重要的作用。因此,粘粒组合中各个粘粒之间发生同源重组的位置对病毒拯救的效率至关重要。粘粒是最近数年来才广泛用于基因文库构建的一种新型载体,它是以bac为骨架,除基于f因子的单拷贝复制子ori2外,还有一个可诱导的oriv高拷贝复制起始点,可诱导达到高拷贝(10~200个)。单拷贝的复制子可减少文库dna片段的重组,使外源dna片段保持稳定;诱导型高拷贝oriv则保证在诱导物存在的情况下下游工作的顺利进行;用其构建的文库保真性和稳定性较好(magriniv,warrenwc,wallisj,goldmanwe,xuj,mardiser,mcphersonjd.fosmid-basedphysicalmappingofthehistoplasmacapsulatumgenome.genomeres.2004,14:1603-1609;zhangl,baoz,chengj,lih,huangx,wangs,zhangc,huj.fosmidlibraryconstructionandinitialanalysisofendsequencesinzhikongscallop(chlamysfarreri).marbiotechnol(ny).2007,9:606-612.)。
本发明从致细胞病变(cpe)出现的时间与第一代拯救病毒的滴度对10组粘粒组合拯救病毒的效率进行了比较,进而确定发生同源重组的最佳位置。
本发明将基因组dna片段末端补平修饰后取30~48kb之间的dna与pcc1fos载体连接,经过末端测序分析,共获得插入外源片段的粘粒200个。
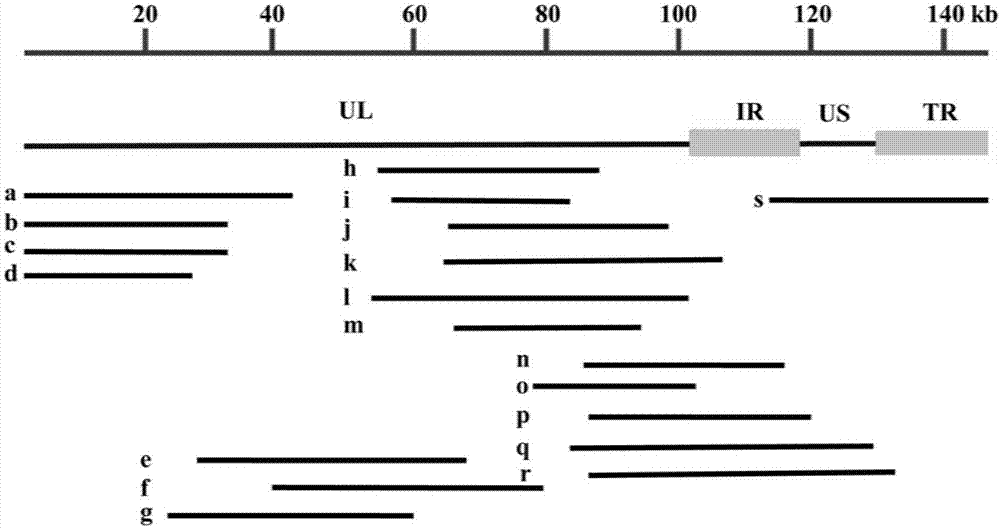
本发明从这200个克隆中筛选出基因组插入片段大小为30-45kb的19个粘粒用于筛选,从这19个覆盖prv全基因组的克隆中选取5种粘粒分10种组合用于拯救病毒,每组组合中粘粒上插入的prvdna片段的两端都能相互重叠,并且5个粘粒能够覆盖prv全基因组。
将10种组合的粘粒分别转染vero细胞,分别观察转染后出现典型cpe的时间,检测拯救病毒的毒价,进而筛选出拯救病毒的最佳组合。
本发明将10种组合的粘粒分别转染vero细胞后,其中,分组1、分组3、分组4、分组5和分组6这5种粘粒组合转染后54h内出现cpe。将病变细胞反复冻融,上清接种pk-15细胞,均能产生典型的cpe,表明病毒拯救成功。ifa结果显示,拯救的病毒与亲本病毒均能与prv阳性血清反应。
这5种粘粒组合的每一个粘粒在prv基因组中的相对位置(nt)如下:
第ⅰ种组合(表2中的分组1的5个粘粒组合b+e+h+p+s);第1个粘粒fostj-b,其在prv基因组的位置是1-35363nt;第2个粘粒fostj-e,其在prv基因组的位置是29544-67498nt;第3个粘粒fostj-h,其在prv基因组的位置是55457-87000nt;第4个粘粒fostj-p,其在prv基因组的位置是82459-124194nt;第5个粘粒fostj-s,其在prv基因组的位置是113582-143642nt。
第ⅱ种组合(表2中的分组3的5个粘粒组合c+g+l+r+s):第1个粘粒fostj-c,其在prv基因组的位置是1-32129nt;第2个粘粒fostj-g,其在prv基因组的位置是23721-59498nt;第3个粘粒fostj-l,其在prv基因组的位置是49114-89118nt;第4个粘粒fostj-r,其在prv基因组的位置是86680-125105nt;第5个粘粒fostj-s,其在prv基因组的位置是113582-143642nt。
第ⅲ种组合(表2中的分组4的5个粘粒组合a+f+o+q+s);第1个粘粒fostj-a,其在prv基因组的位置是1-41633nt;第2个粘粒fostj-f,其在prv基因组的位置是37942-69996nt;第3个粘粒fostj-o,其在prv基因组的位置是62988-100570nt;第4个粘粒fostj-q,其在prv基因组的位置是81316-117252nt;第5个粘粒fostj-s,其在prv基因组的位置是113582-143642nt。
第ⅳ种组合(表2中的分组5的5个粘粒组合b+e+h+q+s):第1个粘粒fostj-b,其在prv基因组的位置是1-35363nt;第2个粘粒fostj-e,其在prv基因组的位置是29544-67498nt;第3个粘粒fostj-h,其在prv基因组的位置是55457-87000nt;第4个粘粒fostj-q,其在prv基因组的位置是81316-117252nt;第5个粘粒fostj-s,其在prv基因组的位置是113582-143642nt。
第ⅴ种组合(表2中的分组6的5个粘粒组合b+e+i+q+s):第1个粘粒fostj-b,其在prv基因组的位置是1-35363nt;第2个粘粒fostj-e,其在prv基因组的位置是29544-67498nt;第3个粘粒fostj-i,其在prv基因组的位置是48838-83227nt;第4个粘粒fostj-q,其在prv基因组的位置是81316-117252nt;第5个粘粒fostj-s,其在prv基因组的位置是113582-143642nt。
本发明经多次重复试验发现,第ⅲ种粘粒组合(即表2中的分组4)转染36h后可见vero出现prv引起的典型病变,其它几种粘粒组合转染54h后可见vero出现典型病变。此外,第ⅲ种粘粒组合拯救病毒的病毒滴度最高,显著高于其它几种组合所拯救病毒的滴度。该组拯救病毒的病毒粒子形态以及生长特性与亲本病毒一致,因此,第ⅲ种粘粒组合拯救病毒的效率最高。第ⅲ种粘粒组合的5个粘粒跨越了prv基因组的ul区和us区,该组合的前三个粘粒(fostj-a、fostj-f、fostj-o)覆盖了基因组的ul区,第四个粘粒(fostj-q)包括全部ir序列以及部分ul区与us区的基因,第五个粘粒(fostj-s)包含了全部us区以及全部tr序列的基因。
本发明fosmid文库的构建方法操作步骤简单,大大缩短了构建文库的周期;与传统的构建文库的载体相比,粘粒具有较高的稳定性。本发明构建方法将巨大的基因组分成几个小片段构建文库提高了文库的稳定性,同时,使基因的改造和修饰更加方便。
本发明进一步利用所构建的fosmid文库结合red/et重组技术快速构建了一株能够稳定融合表达增强型绿色荧光蛋白(enhancedgreenfluorescentprotein,egfp)基因的报告病毒rprvtj-vp26-egfp;经检测,所拯救的报告病毒rprvtj-vp26-egfp与亲本病毒的滴度无统计学差异,生长特性一致。
传统的构建prv突变体的方法主要包括基于同源重组技术构建重组病毒以及基于bac文库与red/et重组技术构建重组病毒。但是,同源重组技术构建重组病毒存在诸多缺点,例如:构建带有左右同源臂的转移载体耗时费力;同源重组效率低;病毒的纯化需要较长时间。prv基因组可高达143kb,极其容易断裂,一旦基因组断裂,便失去了感染性,无法获得重组病毒。以上prv自身的特性给基于bac文库的重组技术带来了极大的困难。crispr/cas9技术提高了构建prv重组病毒的效率。但是,应用该技术同样需要构建1kb左右的同源臂,由于prv基因组g+c含量高达70%,不但影响同源臂的构建,还会影响同源重组的效率,此外,应用该方法也需要进行病毒的纯化。
本发明应用prv多片段拯救系统平台与red/et重组技术构建重组病毒过程中单独操作40kb左右的粘粒不会发生基因组的断裂;同源重组过程只需要50bp的同源臂,可以直接与引物一起合成,节约了转移载体构建的时间;改造粘粒与其它粘粒共同转染拯救重组病毒,重组病毒不需要纯化。以本发明所构建的fosmid文库为基础的多片段拯救重组病毒平台显著地提高了获得重组病毒的效率,可以在14天内构建出prv突变体,而应用同源重组的方法构建重组病毒需要40天左右。因此,基于本发明所构建的prvfosmid文库也为其他dna病毒突变体的构建提供了一个良好的解决办法,具有较为重要的科学意义。
应用同源重组方法、prvbac文库以及crispr/cas9技术获得突变病毒均存在一些缺陷。例如:文库构建周期长、重组病毒拯救效率低、重组病毒纯度低。本发明建立的prv多片段拯救系统克服了以上缺点,将为以prv-tj株为载体构建重组疫苗及相关基础研究奠定了坚实的基础,因此,本发明具有很高的应用价值。
总之,本发明成功构建了prvtj株的fosmid基因文库,从中选取了10组能覆盖完整prv基因组且相互有重叠区域的5粘粒系统,用其转染vero细胞,从中筛选出5组可拯救出完整prv的系统,还根据拯救病毒的效率及特性筛选出拯救病毒的最佳粘粒组合;同时,应用该粘粒组合快速地构建了与vp26融合表达egfp的prv-tj株。检测结果显示,所拯救的报告病毒与亲本病毒在滴度与生长特性上无统计学差异,表明该平台适用于prv重组病毒的快速构建,为今后对prv基因组进行快速、准确修饰以及构建重组疫苗奠定了重要的基础。
附图说明
图1prv-tj株基因组及多片段拯救系统各粘粒相对位置;a、b、c等字母与表2中的粘粒相对应,分别代表不同的粘粒。
图2粘粒(1-41633)的修饰与融合表达egfp基因的重组病毒的构建策略;a:prv基因组结构以及粘粒(1-41633)的基因组位置;b:fosmid-rpslneo与fosmid-egfp构建策略。
图3拯救病毒对细胞的感染及间接免疫荧光试验鉴定。
图4拯救病毒的鉴定;a:拯救病毒的pcr鉴定;b:拯救病毒的病毒粒子形态观察;c:拯救病毒酶切鉴定;d:拯救病毒与prv-tj株病毒生长曲线。
图5rprv-tj-vp26-egfp的拯救与鉴定;a:拯救病毒的cpe;b:拯救病毒的pcr鉴定。
具体实施方式
以下结合具体实施例来进一步描述本发明,本发明的优点和特点将会随着描述而更为清楚。但这些实施例仅是范例性的,并不对本发明的范围构成任何限制。本领域技术人员应该理解的是,在不偏离本发明的精神和范围下可以对本发明的细节和形式进行修改或替换,但这些修改和替换均落入本发明的保护范围内。
试验例伪狂犬病病毒基因组fosmid文库的构建、最佳组合的粘粒筛选及伪狂犬病病毒突变体的快速构建
1材料和方法
1.1主要试剂
prv变异株tj株(prv-tj)由本发明人实验室分离、鉴定和保存;非洲绿猴肾细胞(vero细胞)、猪肾细胞(pk-15细胞)由本发明人实验室保存,用含有10%胎牛血清(fbs)的dmem培养基(gibco)于37℃、5%co2环境中培养;大肠杆菌dh5α感受态细胞由本发明人实验室制备和保存;pok12载体质粒购买于novagene公司,pegfp-n1载体质粒购买于clontech公司,pmd18-tsimplevector质粒购于宝生物(takara)公司;fitc标记的羊抗猪igg荧光二抗购买于sigma公司。
copycontrolfosmidlibraryproductionkit(ccfos110)、pcc1fostmfosmidvector、epi300tm-t1re.coliplatingstrain、包装蛋白maxplaxtmlambdapackagingextracts均购自epicentre公司;kpni、ncoi和psti限制性内切酶购自takara公司;低熔点琼脂糖购自invitrogen公司(ultrapuretmlmpagarose,16520-050),所有抗生素与l-阿拉伯糖(sigma,a-3256)均购自sigma公司;质粒小提试剂盒购自epigenetics公司(zrbacdnaminiprepkit,d4049);质粒中提试剂盒购自qiagen公司(qiafilterplasmidmidikit,12243);x-tremegenehpdna转染试剂购自roche公司(cat.no.06366546001)。
1.2prv基因组的提取
参照pomeranz等的方法进行prv基因组的提取(pomeranzle,reynoldsae,hengartnercj.molecularbiologyofpseudorabiesvirus:impactonneurovirologyandveterinarymedicine.microbiol.mol.biol.rev.2005,69:462-500.)。简述如下:将病毒接种于pk-15细胞,当90%细胞出现cpe时,收集细胞于50ml离心管。使用10mllcm溶液裂解细胞。加入1ml氟利昂(1,1,2-三氟三氯乙烷),充分震荡,释放病毒粒子。超速离心机离心2h,4℃26000r/min,加入10mlten与500μl10%sds释放病毒基因组。使用等体积的平衡酚溶液抽提一次,酚:氯仿:异丙醇(25:24:1)溶液抽提二次。然后用冷无水乙醇沉淀dna,最后用去离子水溶解基因组。
1.3prvfosmid文库的构建
取提取的基因组dna,应用物理法随机剪切,按照copycontrolfosmidlibraryproductionkit的使用说明对dna进行末端补平,并进行低熔点琼脂糖凝胶脉冲电泳,然后切下36~48kb之间的dna区域,并回收dna。取0.25μg回收纯化的dna连接到pcc1fostm上,包装、转导至epi300宿主细胞中,将转导产物均匀涂布于含12.5μg/ml氯霉素的lb平板上,37℃过夜培养。随机挑取50个单克隆,提取质粒,并进行末端测序鉴定。将测序正确的粘粒,进行序列拼接,获得涵盖prv全长的fosmid文库。
1.4病毒的拯救
按照x-tremegenehpdna转染试剂说明书,将纯化好的粘粒分成10个组合,然后转染至密度为90%的10cm细胞培养板vero细胞中,每种粘粒的转染剂量为2μg,每个组合含有5种粘粒。待cpe后反复冻融细胞,收获病毒,将拯救的病毒命名为reprv-tj。
1.5间接免疫荧光试验(ifa)
将拯救病毒接种密度为90%的pk-15细胞,48h后用抗prv的多克隆抗体进行ifa实验。具体操作如下:弃掉培养液,用pbs清洗细胞2遍,然后用冷无水乙醇固定细胞,30min后加入猪源抗prv抗体,在室温下作用2h后用pbs洗涤细胞5次,并加入100倍稀释的fitc标记的羊抗猪igg,于室温中避光作用1h,用pbs洗涤细胞5次,5min/次,在倒置荧光显微镜下观察并拍照。
1.6pcr
将拯救的病毒于-80℃/37℃条件下反复冻融后,在pk-15细胞上连续传20代,提取每个毒株第20代病毒的dna,应用特异性引物扩增gb、ge片段,并对扩增片段进行测序分析。
1.7电镜观察
将收获的reprv-tj病毒粒子3000r/min离心10min,取上清液置于新的1.5ml离心管中,13000r/min离心10min。样品经2%磷钨酸负染后,病毒粒子的形态通过透射电子显微镜检测。
1.8脉冲场电泳
按照1.2的方法提取reprv-tj、prv-tj基因组,然后分别使用kpni、ncoi和psti将10μg基因组进行酶切反应,酶切产物进行脉冲场核酸凝胶电泳分析。
1.9病毒拯救的最佳粘粒组合的筛选
经过末端测序分析,共获得插入外源片段的粘粒200个。从这200个克隆中筛选出基因组插入片段大小为30-45kb的19个粘粒用于后续研究。选取10组5粘粒组合用于拯救病毒。其中每组组合中粘粒上插入的prvdna片段的两端都能相互重叠,并且5个粘粒能够覆盖prv全基因组(表3)。将10种组合的粘粒分别转染vero细胞,分别观察转染后出现典型cpe的时间,检测拯救病毒的毒价,进而筛选出拯救病毒的最佳组合。
表1prvtj株基因组及多片段拯救系统各粘粒插入的片段的大小以及基因组的相对位置
表2拯救病毒的粘粒组合
注:a、b、c等字母与表2中的粘粒相对应,分别代表不同的粘粒。
1.10一步生长曲线
将拯救病毒以感染复数(multiplicityofinfection,moi)为10的剂量接种于生长至单层的pk-15细胞,并在感染后0h、4h、8h、12h、16h、20h、24h、28h、32h、36h、48h、60h和72h收获病毒。将不同时间点收获的病毒分别用ifa测定病毒滴度,并绘制出病毒的生长曲线。
1.11融合表达egfp的重组病毒的构建
根据genbank上发表的prvtj株全长基因组序列(accessionno.kj789182)、fosmid序列与egfp基因序列设计了引物(表3)。应用red/et重组技术,经过两步法重组分别构建融合表以下达egfp的重组病毒。
具体操作如下:1)将两端带有50bp大小左、右同源臂的选择标记基因rpsl-neo表达盒插入相应粘粒的vp26基因的终止密码子前方;2)用两端带有同样的50bp左、右同源臂的egfp基因替换选择标记基因rpsl-neo表达盒;3)在含有氯霉素和链霉素双抗性的平板上筛选阳性克隆,经卡那霉素抗性反向筛选和pcr进一步鉴定粘粒;4)提取并纯化改造后的粘粒,转染vero细胞以拯救重组病毒。具体构建策略见图2。
表3red/et重组过程中所用到的引物
2试验结果
2.1fosmid文库构建与鉴定
通过物理剪切法获得随机断裂的prvdna片段;剪切片段末端、补平修饰后与pcc1fos载体连接;用噬菌体包装蛋白包装重组dna,感染大肠杆菌epi300,随机选取单个克隆,提取粘粒,经末端测序证实,共获得了插入外源片段的粘粒200个,分析表明,200个粘粒覆盖了全prv基因组。但其中包含ul区片段的粘粒相对偏多,说明文库存在一定的偏嗜性。基因组插入片段从30kb到45kb不等,共选取了19个能够覆盖全基因组的克隆进行随机组合(表1、表2、图1)。
2.2病毒的拯救
从19个覆盖prv全基因组的克隆中选取10种组合(表3),每种组合含有5个粘粒,每个粘粒的prvdna片段两端都能相互重叠,5个粘粒能够覆盖prv全基因组。将10种组合的粘粒分别转染vero细胞后,其中有5种组合的粘粒转染后48h内出现cpe。将病变细胞反复冻融,上清接种pk-15细胞,均能产生典型的cpe,表明拯救病毒成功。ifa结果显示,拯救病毒与亲本病毒均能与prv阳性血清反应(图3)。
2.3拯救病毒的鉴定
拯救病毒经过20轮传代后,提取病毒dna,经pcr方法均能检测prvge、gb基因(图4a)。透射电子显微镜下观察病毒粒子形态结果表明,拯救病毒具有成熟的病毒粒子,并且可以观察到其囊膜结构,进一步证明成功获得了prv病毒粒子(图4b)。脉冲场核酸凝胶电泳结果显示,拯救病毒与prv-tj株基因组的酶切图谱基本一致(图4c)。此外,拯救病毒与prv-tj株病毒生长曲线没有明显区别(图4d)。
2.4病毒拯救的最佳粘粒组合
经多次重复,第4组粘粒组合转染36h后可见vero出现prv病毒典型病变,其它粘粒组合转染54h后可见vero出现典型病变。第4组粘粒组合拯救病毒的病毒滴度最高,如表3所示。此外,该组拯救病毒的病毒粒子形态以及生长特性与亲本病毒一致。因此,第4组组合拯救病毒的效率最高。第4组粘粒组合包括5个粘粒,这5个粘粒跨越了prv基因组的ul区和us区,具体位置如下:fostj-a:1-41633;fostj-f:37942-69996;fostj-o:62988-100570;fostj-q:81316-117252;fostj-s:113582-143642。该组合的前三个粘粒(fostj-a、fostj-f、fostj-o)覆盖了基因组的ul区,第四个粘粒(fostj-q)包括全部ir序列以及部分ul区与us区的基因,第五个粘粒(fostj-s)包含了全部us区以及全部tr序列的基因。
表4拯救病毒的粘粒组合以及拯救病毒效率的比较
注:a、b、c等字母与表2中的粘粒相对应,分别代表不同的粘粒。
2.5重组病毒rprvtj-vp26-egfp的体外生物学特征
为了检测此fosmid文库在快速构建prv突变体中的应用效果,本试验构建了与vp26基因融合表达egfp报告基因的粘粒,并拯救了表达egfp的重组病毒rprvtj-vp26-egfp,阳性对照组转染粘粒组合4,能够产生cpe,阴性对照组只转染粘粒c、g、l与r,不能够产生cpe(图5)。同时比较了rprvtj-vp26-egfp与prv-tj的生长复制特性,结果显示,rprvtj-vp26-egfp与prv-tj的生长特性一致。
sequencelisting
<110>中国农业科学院哈尔滨兽医研究所(中国动物卫生与流行病学中心哈尔滨分中心)
<120>伪狂犬病病毒基因组fosmid文库、构建方法及其在构建突变体中的应用
<130>hlj-2001-180112a
<160>6
<170>patentinversion3.5
<210>1
<211>74
<212>dna
<213>abedusherberti
<400>1
gcgcgcgggggcgcgcacagacgcgcgctccccgccgagccatcatgtccggcctggtga60
tgatggcgggatcg74
<210>2
<211>74
<212>dna
<213>artificalsequence
<400>2
gcgccctcgagcgtctgcgcggtgatcgtccggggattgttcgggtcgaatcagaagaac60
tcgtcaagaaggcg74
<210>3
<211>69
<212>dna
<213>artificalsequence
<400>3
gcgcgcgggggcgcgcacagacgcgcgctccccgccgagccatcatgtccgtgagcaagg60
gcgaggagc69
<210>4
<211>70
<212>dna
<213>artificalsequence
<400>4
gcgccctcgagcgtctgcgcggtgatcgtccggggattgttcgggtcgaatctagatccg60
gtggatcccg70
<210>5
<211>18
<212>dna
<213>artificalsequence
<400>5
catcatcctgaacatgcg18
<210>6
<211>19
<212>dna
<213>artificalsequence
<400>6
gctgctgtagtcgctggtg19
- 还没有人留言评论。精彩留言会获得点赞!