一种灵敏快速检测金黄色葡萄球菌肠毒素B的检测方法与流程
本发明涉及食品安全检测分析技术领域,具体而言,涉及一种灵敏快速检测金黄色葡萄球菌肠毒素b的检测方法。
背景技术
金黄色葡萄球菌是引起细菌性食品中毒的重要食源性致病菌,广泛存在于动物的体表、人的鼻腔及腐败的食物中,其本身并无致病性,但是其分泌肠毒素等细胞外毒素能够引起中毒反应。其中以金黄色葡萄球菌肠毒素b(staphylococcalenterotoxinb,seb)为例,具有耐热、耐低ph、耐胰蛋白酶分解的能力,误食或者吸入体内会引起引发恶心、呕吐、腹部痉挛等中毒现象。由于制作成本低、污染面积大,传播方式多样,seb被美国cdc列为b类潜在的生物战剂,同时也是联合国《禁止生物武器公约》中明确规定的重要生物战剂。
杂交链式反应(hcr)在生物传感领域广泛用作核酸信号放大,相比于传统的聚合酶链式反应(pcr)等方法,hcr方法成本低,并且对环境具有很好的抗干扰能力。目前,传统的基于hcr生物传感检测方法已应用于比色、电化学、荧光等多种检测平台,其中比色法虽然能够直观显示检测结果,但在实现精准定量方面较为困难。电化学方法需要反复的结合和洗脱过程,这在一定程度上增加了分析样品检测步骤,提高了检测时间。
seb核酸适配体是一段采用selex富集获得的能够识别seb的单链dna,它和抗体一样具有优越的特异性识别能力,是目前在生物传感领域逐渐广泛使用的一类分子识别元件,因此仅需要采用化学合成的方式就可以获得成本低、批间稳定性较高的seb特异性识别元件。最初将分子信标引入到核酸适配体主要是基于核酸分子杂交的分子信标检测方法,这种设计需要将核酸适配体分为两个部分,其中一部分设计成分子信标的结构,当目标物存在的条件下,从而引起核酸分子构象的变化,从而产生荧光“恢复”或者“猝灭”。上述方法能够实现的前提是该适配体具有独特的二级结构,但目前大部分的核酸适配体结构上难以实现上述功能,这在一定程度上限制了分子信标在核酸适配体上的应用。
技术实现要素:
本发明目的在于提供了一种灵敏快速检测金黄色葡萄球菌肠毒素b的检测方法,与传统基于核酸适配体“分子开关”的检测技术相比检测灵敏度降低2个数量级。
本发明的目的通过如下技术方案实现:
一种灵敏快速检测金黄色葡萄球菌肠毒素b的检测方法,其特征在于,具体步骤如下:
(1)反应前将浓度为5μm的h1和h2于95℃下加热变性5min,然后室温下缓慢降温1h,1μmseb核酸适配体于85~95℃下加热变性5~10min,之后在快速降温并于冰浴条件下保持15min左右,以上所述的寡核苷酸链的名称及序列如下表;
(2)取浓度为1μmseb适配体6.7-33.3μl,加入seb100μl,混合后加入80-100μl的杂交缓冲液,37℃于恒温混匀仪700rpm下反应30min;
(3)加入1μmh06.7-33.3μl,37℃于恒温混匀仪700rpm下反应30min;
(4)加入浓度为5μmh1100μl和浓度为5μmh2100μl混匀,配制成混合反应体系,每试管中加入量为10-30μl;
(5)将该反应体系置于37℃恒温混匀仪700rpm下反应45min,测定荧光度值,参数设置条件如下:激发波长488nm,发射起始波长510nm,发射终止波长600nm,扫描速度1000nm/min,扫描间隔1nm,等待时间0s,激发带宽10nm,发射带宽10nm增益(pmt):高900v。
作为本发明更优的技术方案:所述的步骤(2)中的1μmseb适配体20-30μl。
作为本发明更优的技术方案:所述的步骤(3)中的1μmh0为20-30μl。
作为本发明更优的技术方案:所述的步骤(2)中的1μmseb适配体20μl,所述的步骤(3)中的1μmh0为30μl,所述的seb适配体与h0的添加比例为1:1.5。
作为本发明更优的技术方案:所述的步骤(4)中的配制成混合反应体系,每试管中加入量为20μl。
本发明探索了不同类型的发卡dna探针,通过dna探针的序列设计、软件模拟和电泳实验表征该序列能够实现稳定的恒温扩增过程,并同时针对不同探针浓度分别进行优化,最终采用竞争法杂交链式反应实现对seb的特异性检测。其中发卡h1设计成类分子信标的形式,由于传统基于单一分子信标所产生的信号强度有限,通过与hcr方法的结合从而释放更多荧光信号。该方法灵活巧妙,这种无酶的扩增方法应用于复杂样品基质中具备良好的抗干扰优势,而且较短的扩增时间符合现场检测的需求。相比于传统基于核酸适配体“分子开关”的检测技术,该方法的检测灵敏度降低2个数量级,所以本发明对肠毒素b的快速检测具有重要的实际意义,对于实现现场快速检测技术具有很好的指导意义。通过该方法可以特异性定量检测牛奶中seb的含量,其中通过构建基于seb核酸适配体的分子开关,并以其部分互补的cdna序列作为引发链,稳定存在于体系的h1经过荧光基团和猝灭基团标记,当h1以发卡结构形式存在时,由于荧光共振能量转移的作用使得荧光猝灭。当seb存在时,cdna与seb竞争性结合seb适配体,seb浓度越高,cdna量越高,从而引发h1和h2循环扩增的能力越强,荧光恢复值越高。
附图说明
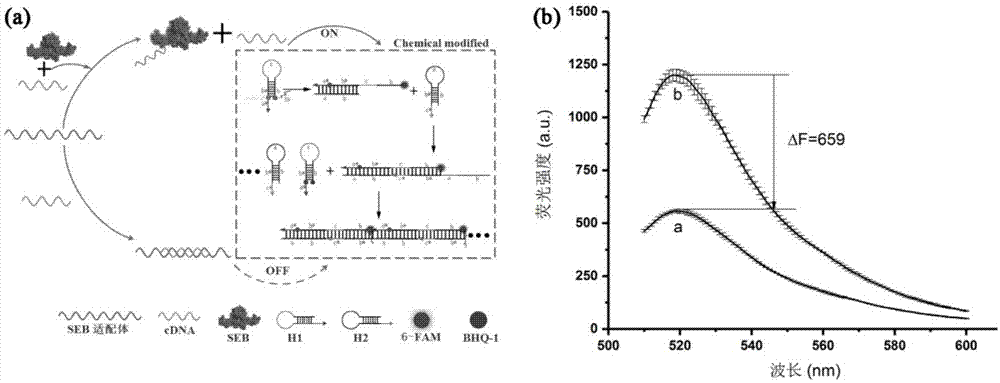
图1(a)荧光杂交链式反应用于seb检测的原理图;(b)发卡dna快速及慢速变复性的荧光光谱图比较;
图2表征确证竞争荧光杂交链式反应检测seb体系凝胶电泳图;
图3荧光法测定hcr动力学过程标准曲线图;
图4aptamer/h0的比例优化曲线图;
图5h1和h2总添加体积(1:1)的优化曲线图;
图6竞争荧光杂交链式反应体系应用于检测不同浓度的seb的荧光光谱图(a)及标准曲线图(b);
图7传统荧光核酸适配体竞争法检测seb标准曲线图。
具体实施方式
本发明在传统核酸适配体构建荧光“分子开关”的基础上,引入hcr核酸等温扩增进一步降低检测的灵敏度,该竞争法hcr信号放大检测seb的具体原理如图1(a)所示。其中部分互补链序列(cdna)是基于seb核酸适配体序列的中间位置进行设计。在体系中不含seb的情况下,cdna通过碱基互补配对与seb核酸适配体杂交,相反,cdna将会游离在检测体系中。同时,以cdna(h0)为基础设计发卡结构序列h1和h2。线性hcr系统包含两个发夹底物h1和h2和单链dna(h0)。利用nupack和mfold软件设计两个发夹探针h1和h2并模拟验证杂交反应过程。首先,h1中的6nt(a*)粘性末端及其相邻的茎(b*)与h0配对。然后,h1的6nt(c)和18nt(b)的茎环将与h2杂交并打开h2的茎部,发卡h1和h2通过碱基互补配对产生的熵自由能持续催动杂交配对结合,从而产生一条长链h0-(h1-h2)n复合物。h1在其5'末端的6-fam标记,与发夹结构中的bhq-1猝灭剂对应的位置上。邻近的荧光团和猝灭剂可以通过荧光共振能量转移(fret)猝灭6-fam的荧光。相反,当h1被打开时,荧光基团和猝灭基团远离,荧光值恢复。因此,不同浓度的cdna(h0)会引发不同的荧光信号强度,以此对目标物seb进行间接定量。
下面将结合实例对本发明的实施方案进行详细描述,实例中未注明的具体条件者,皆按照常规条件或制造商建议的条件进行,使用中适配体序列,cdna序列及发卡h1和h2序列均由上海生工生物(北京合成部)合成,实验中所用到的肠毒素标准品(1mg/ml)是由sigma公司购买,genegreen染料购置天根生化科技有限公司,f97pro荧光分光光度计购自上海棱光技术有限公司,imagequant350凝胶成像系统购自gehealthcare。
实施例1:发卡探针的设计及变复性条件的确定
为了保证序列设计的合理性,发卡探针运用nupack软件设计,在analysis的工作界面中,调整核酸的类型为dna,工作温度设置为37℃,同时在advancedoptions选项框中,设置na+为0.4m,mg2+为5mm。首先使用nupack软件验证设计的h1和h2序列能否正常形成发卡结构。其次,设置numberofstrandspecies为2,maxiumcomplexsize为4,将h1和h2以等浓度进行杂交运算,验证h1和h2在无引发链h0的条件下有无杂交。最后,设置numberofstrandspecies为3,maxiumcomplexsize为5,其他参数不变的条件下,将h0或sebaptamer的序列分别导入到软件中去,同时引入h1和h2的序列见表1和表2,验证有无非特异扩增及能否以正常的形式扩增。实验设计的cdna、发卡探针h1和h2干粉先用tris-hcl(浓度为20mm,ph7.2)储备缓冲液溶解,然后用杂交缓冲液(tris-hcl浓度为20mm,nacl浓度为400mm,ph7.2)将发卡探针h1和h2配成5μm。图1b(a)为1号pcr管至于pcr仪中进行缓慢变复性处理,设置参数为95℃,10min;85℃,10min;75℃,10min;65℃,10min;55℃,10min;45℃,10min;35℃,10min;25℃,60min;与此同时,图1b(b)2号pcr管中已经变性的dna迅速置于冰浴条件下降温10min,取出,备用。将以上两种复性方式分别于荧光分光光度计上测定其荧光强度。图1(b)中表明使用慢速复性的发卡h1的荧光强度比快速复性发卡dna的荧光强度值低659,此时更有利于后续检测过程控制阴性值到最低水平,故后期实验中发卡h1和h2采用慢速复性处理:95℃,10min;85℃,10min;75℃,10min;65℃,10min;55℃,10min;45℃,10min;35℃,10min;25℃,60min。
实施例2:凝胶电泳表征确证竞争荧光杂交链式反应检测seb体系可行性
(1)发卡dnah1和h2及seb核酸适配体的预处理:反应前将浓度为5μm的h1和h2于95℃下加热变性5min,之后在缓慢降温并于4℃条件下保持1h,1μmseb核酸适配体于95℃下加热变性5min,然后在冰水浴条件下快速降温。
(2)凝胶电泳表征验证hcr扩增:在tris-hcl缓冲液(20mm,400mmnacl,5mmmgcl2,ph7.2)中分别加入10μlh1(5μm)、10μlh2(5μm)、引发链10μl(0、0.63、1.25、2.5、5、10μm),将反应溶液混匀后在金属浴中37℃恒温反应60min。
(3)凝胶电泳表征验证竞争荧光杂交链式反应检测seb:取浓度为1μmseb适配体20μl,加入10ng/mlseb10μl,混合后加入1μm引发链10μl并于37℃下震荡反应1h.反应结束后加入10μlh1(5μm)、10μlh2(5μm),37℃下恒温反应60min。
(4)琼脂糖电泳:称取约2g琼脂糖100ml1×tae电泳缓冲液中,在微波炉中反复加热、摇匀至琼脂糖完全溶解。待温度逐渐降低时,加入10μlgenegreen染料混匀。紧接着进行灌胶、固定、加样后,经120v电压分离60min于荧光成像仪进行拍照。
(5)非变性聚丙烯酰胺(page)电泳
a.装好灌胶装置,配制12%的page凝胶,加入胶板中使用梳子插入到凝胶中固定。
b.将凝胶板固定在电泳槽上。用1*tbe缓冲液注满上层及下层槽,预先电泳400v,30min预热。临加样前用移液枪上下吹打tbe缓冲液数次以彻底洗涤样品孔。
c.加样:加入等体积2xdna非变性缓冲液20μl并与之混匀。
d.电泳及染色:80v下1.5h后取出胶片于genengreen染色液中染色1h。
e.将凝胶取出放在塑料膜上,使用iq350凝胶成像系统拍照。
如图2(a)所示,其中孔道1和孔道10分别是50bpdnaladderandd2000marker,孔道2、3和9分别是发卡h1、h2和h1与h2的混合体系。其中孔道9中仅为一条明亮的条带,说明在没有h0的条件下,hcr未出现非特异性扩增。孔道4-8分别为添加不同浓度的h0对于扩增的影响,结果表明当h0浓度较高时,hcr的扩增长度相对集中在较短的碱基长度区间内,当h0逐渐由3.3μm降低至0.42μm时,hcr扩增的平均长度变高。当继续降低时,扩增的效率范围降低。图2(b)是将相同样品加样于非变性丙烯酰胺电泳上,其电泳效果同前者。图2(c)表征了基于seb适配体的hcr检测体系。泳道2显示了加入h0后引发hcr扩增。泳道3为在不含seb的检测体系(seb适配体+h0+h1+h2),并且仅出现两条,一条为seb适体的碱基,另一条为h1和h2的混合物。然而,当添加seb时,条带的增加说明可以引发hcr扩增(泳道4)。另外,泳道5显示单纯seb适配体不能触发hcr扩增,以上结果表明该检测体系具有良好的特异性。
实施例3:荧光法测定hcr动力学过程
(1)反应前将浓度为5μm的h1和h2于95℃下加热变性5min,之后在缓慢降温并于4℃条件下保持1h,其中所使用序列参见表1。
(2)配制反应体系,取1136μl的杂交缓冲液,加入80μlh1(5μm)和80μlh2(5μm)混匀,配制成混合反应体系。
(3)取20μl浓度为10μm的引发链h0,分别稀释至5μm、2.5μm、1.25μm、0.625μm、0.3125μm,每个浓度至少10μl。
(4)将该反应体系至于37℃恒温水浴锅中,避光反应2h。
由于h1设计成类分子信标的形式,当发卡结构被打开时,荧光发生恢复,此时为了研究在不同h0存在的条件下,与此对应的荧光值大小是否存在线性或者非线性动力学规律。分别在0.0313-1μm的范围内其荧光强度恢复的大小情况,如图4.5所示,荧光强度随着h0的浓度增加而逐步增高。且荧光强度与log2c(h0)成明显的线性关系。经线性拟合结果表明,y=1059.40log2x+1995.45,r2=0.9808。该动力学模型为初步建立hcr检测方法提供了理论依据。
实施例4:不同aptamer/h0的添加对hcr反应的影响
(1)反应前将浓度为5μm的h1和h2于95℃下加热变性5min,然后室温下缓慢降温1h,1μmseb的核酸适配体于85~95℃下加热变性5~10min,之后在快速降温并于冰浴条件下保持15min左右,其中所使用序列参见表1。
(2)sebaptamer与seb的结合反应,分别取浓度为1μmsebaptamer33.3、32、26.7、20、13.3、8、6.7μl,加入不同稀释浓度的seb100μl,混合后加入80μl的杂交缓冲液,37℃于恒温摇床(200rpm)下反应1h。
(3)与此对应分别加入h06.7、8、13.3、20、26.7、32、33.3μl,37℃于恒温摇床200rpm下反应1h。
(4)加入浓度为5μmh1100μl和浓度为5μmh2100μl混匀,配制成混合反应体系,每试管中加入量为20μl。
(5)将该反应体系置于37℃恒温混匀仪700rpm下反应45min。测定荧光度值。参数设置条件如下:激发波长488nm,发射起始波长510nm;发射终止波长600nm;扫描速度1000nm/min;扫描间隔1nm;等待时间0s;激发带宽10nm;发射带宽10nm增益(pmt):高900v。
结果如图4表明,在seb的浓度固定的条件下,aptamer/h0比例越高,表明seb与seb适配体的结合量在保持不变的条件下,h0将更多的与游离的seb核酸适配体发生杂交,从而引起h0的浓度相对较低,无法产生较为充分的hcr扩增反应,荧光值较低。aptamer/h0比例越低,h0的浓度相对增加,从而大大提高了荧光检测信号。当aptamer/h0逐渐降低至1:1.5,相对荧光强度基本保持平稳,在底物浓度h1和h2都固定的条件下,继续降低aptamer/h0的比例,此时反应的荧光强度会增加,但是与此同时背景噪声将会大大增加。因此,选择aptamer/h0的最适比例为1:1.5。
实施例5:不同h1和h2添加量对hcr反应的影响
(1)反应前将浓度为5μm的h1和h2于95℃下加热变性5min,然后室温下缓慢降温1h。1μmseb的核酸适配体于85~95℃下加热变性5~10min,之后在快速降温并于冰浴条件下保持15min左右,其中所使用序列参见表1。
(2)取浓度为1μmseb适配体20μl,加入100ngml-1的seb100μl,混合后加入80μl的杂交缓冲液,37℃于恒温混匀仪700rpm下反应30min。
(3)加入1μmh030μl,37℃于恒温混匀仪700rpm下反应30min。
(4)加入浓度为5μmh1100μl和浓度为5μmh2100μl混匀,配制成混合反应体系,每试管中加入量分别为10、15、20、25、30μl。
(5)将该反应体系置于37℃恒温混匀仪700rpm下反应45min。测定荧光度值。参数设置条件如下:激发波长488nm,发射起始波长510nm;发射终止波长600nm;扫描速度1000nm/min;扫描间隔1nm;等待时间0s;激发带宽10nm;发射带宽10nm增益(pmt):高900v。
结果如图5表明,当h1/h2的添加量为25μl时,相对荧光强度达到最大值,这是因为在该条件下,h1和h2杂交反应比较充分,形成较多的长链h1-h2复合结构。考虑到成本等因素,当h1/h2的添加量为20μl时,相对荧光强度并无显著性差异,从而确定h1和h2的最适添加量为20μl。
实施例6:竞争荧光杂交链式反应体系应用于检测不同浓度的seb的荧光光谱图
(1)反应前将浓度为5μm的h1和h2于95℃下加热变性5min,然后室温下缓慢降温1h。1μmseb的核酸适配体于85~95℃下加热变性5~10min,之后在快速降温并于冰浴条件下保持15min左右,其中所使用序列参见表1。
(2)取浓度为1μmseb适配体20μl,加入不同稀释浓度3.13、6.25、12.5、25、50、100、200ngml-1的seb100μl,混合后加入80μl的杂交缓冲液,37℃于恒温混匀仪700rpm下反应30min。
(3)加入1μmh030μl,37℃于恒温混匀仪700rpm下反应30min。当h0添加体积在13.3~30μl时候,相对荧光强度为0.62~0.8;当h0添加体积大于30μl时,相对荧光强度约0.98。
(4)加入浓度为5μmh1100μl和浓度为5μmh2100μl混匀,配制成混合反应体系,每试管中加入量为20μl,当h1/h2的添加量为10~20μl时,hcr反应不完全,此时相对荧光强度为0.61~0.95,当h1/h2的添加量为30μl时,dna杂交分子间可能存在分子位阻,从而限制了反应的进行,此时相对荧光强度为0.80。
(5)将该反应体系置于37℃恒温混匀仪700rpm下反应45min。测定荧光度值。参数设置条件如下:激发波长488nm,发射起始波长510nm;发射终止波长600nm;扫描速度1000nm/min;扫描间隔1nm;等待时间0s;激发带宽10nm;发射带宽10nm增益(pmt):高900v。
根据荧光光谱图6(a)可知,随着seb浓度逐渐增加,体系在520nm的荧光强度逐渐增加。同样在图6(b)所示,以seb浓度的对数为横坐标,体系中测定的荧光强度为纵坐标作图,可得出在3.13-200ngml-1为纵坐标作图,在3.13~100ngml-1的范围内,seb浓度的对数值与对应的荧光强度值具有良好的线性关系,当seb浓度低于3.13ngml-1时,其产生的荧光值接近于阴性值,故无法进行定量;当seb浓度高于100ngml-1时,产生的荧光强度突然提高,这可能是由于seb核酸适配体基本结合完全,导致释放的h0量过高导致,
其中线性方程为y=241.8876log2x+351.9958,r2=0.9902。三倍信噪比(3σ/k)计算出检出限为0.33ngml-1。
实施例7:竞争荧光杂交链式反应体系应用于检测不同浓度的seb的荧光光谱图
(1)反应前将浓度为1μm的sebaptamer1于95℃下加热变性5min后置于冰水浴下快速复性10min。
(2)标记有荧光基团的sebaptamer1与seb的结合反应,取浓度为1μmsebaptamer20μl,加入不同稀释浓度的seb100μl,混合后加入80μl的杂交缓冲液,37℃于恒温混匀仪(700rpm)下反应30min。
(3)加入标记有猝灭基团的1μmcdna120μl,37℃于恒温混匀仪(700rpm)下反应30min。测定样品的荧光强度。
传统的荧光适配体“分子开关”相比在提高seb检测灵敏度的优势,图7为方法学比对的实验结果,从图中可以看出,在31.25-1000ngml-1的范围内,荧光强度较之前有很大的提高。其中线性方程为y=142.453log2x+1732.1943,但是检出限为(3σ/k)6.94ngml-1,高出核酸适配体-杂交链式反应荧光检测方法的21倍,证明hcr在传统荧光适配体的“分子开关”基础上发生了进一步的扩增,符合反应机理,其原理图见图1(a)。
表2寡核苷酸链的名称及序列
其中#代表了修饰有6-fam荧光基团。*代表修饰有bhq-1猝灭基团。
- 还没有人留言评论。精彩留言会获得点赞!