一种邻醛基苯基脂肪酸的产业化合成方法与流程
本发明涉及药物中间体领域,特别涉及一种邻醛基苯基脂肪酸的产业化合成方法。
背景技术:
邻醛基苯基脂肪酸是重要的一类药物中间体,如邻羧基苯甲醛是合成他尼氟酯和酞氨苄青霉素等药物的重要中间体,其具有广泛的市场需求。现有技术中,邻醛基苯基脂肪酸的主要合成方法有:(1)以芳香内酯为原料,以nbs(n-溴代琥珀酰亚胺)或者三溴化磷卤化制得产品,最终产品收率虽然较高(约60%~70%),但缺点是需要制备nbs,且经济效益不佳。其原因在于,nbs或者三溴化磷的用量大,但不能形成溴源闭合循环,流失量大。(2)采用芳香内酯为原料,以溴素卤化制得产品;但该方法产品收率偏低(约30%),且同样存在不能形成溴源闭合循环,溴素用量大,废水排放造成严重环境污染的问题。不论是高成本还是环保压力,抑或是传统工艺造成的低收率,都不可避免的会成为限制邻醛基苯基脂肪酸大规模持续生产的重大问题。
因此,如何获得低成本、高产率、高品质、及绿色环保的邻醛基苯基脂肪酸的产业化工艺仍值得去探索。
技术实现要素:
为了解决上述问题,本发明人进行了锐意研究,结果发现:以芳香内酯或者邻甲基苯基脂肪酸为原料,经卤化反应、水解,得到邻醛基苯基脂肪酸;其中,在一种方式中,卤化反应中的卤族元素来源为卤盐如nabr或者卤化氢溶液如hbr,卤盐或者卤化氢经氧化物活化生成卤素单质后参与卤化反应,由于氧化剂的存在,可源源不断生成卤素单质参与反应,水解后卤族元素由中间体中脱离,再次形成卤盐或者卤化氢,可实现卤素的闭合循环;在另一种方式中,卤化反应中的卤族元素来源为卤素单质,采用与反应原料等当量或者略过量的卤素单质反应,反应后期系统中卤素单质反应(几近)完全,溶剂中存在卤素离子,加入氧化剂,将卤素离子氧化生成卤素单质后参与卤化反应,在实现原料反应完全的前提下,降低了卤素单质用量,同样可实现卤素的闭合循环。本发明从根本上解决了卤素的闭合循环,节省了大量原材料成本,减少环境污染,同时收率较高(位于行业顶尖水平),从而完成了本发明。
本发明的目的在于提供以下方面:
(1)一种邻醛基苯基脂肪酸的合成方法,该方法包括:以芳香内酯或者邻甲基苯基脂肪酸为原料,经卤化反应、水解,得到邻醛基苯基脂肪酸;
所述芳香内酯的结构为:
其中,x选自氢元素或者卤素,所述卤素选自f、cl或者br,优选为f或者cl,更优选为cl;n取值0~2;
所述邻甲基苯基脂肪酸的结构为:
根据本发明提供的一种邻醛基苯基脂肪酸的产业化合成方法,具有以下有益效果:
(1)本发明极大的扩展了工艺过程中传统卤源的选择范围,摈弃了文献报道过的价格昂贵的nbs或者pbr3,或者不易操作的卤素单质,这直接降低了原料的成本和产业化操作难度。
(2)本发明在卤化反应过程中加入设定的氧化剂,通过将反应体系中的卤素离子氧化/活化为卤素单质,使得在采用br2或者i2单质以外的卤源进行反应时,提高卤化反应速率;
同时,氧化剂的加入利于反应原料(芳香内酯、邻甲基苯基脂肪酸)转化率的提高,对此,在产业化合成过程中,能够大幅度降低卤源的用量,利于成本控制、环保以及对溴素、溴化氢等卤源的操作。
(3)氧化剂以滴加或者分多次加入反应体系中,使得卤素单质不存在局部浓度过大的问题,这样有效降低了对苯环上氢的取代造成的副反应增加的问题。
(4)本发明中,在水解反应阶段,采用回流方式进行水解,可极大提高水解反应的效率,且采用较少的水解溶剂即可完成整个水解反应。
(5)本发明中,水解后母液中能够重新产生相应的卤盐或卤化氢溶液,再次套用至卤化反应阶段,解决了产业化合成过程中卤源闭合循环的难题,极大地降低了环保压力,以及卤素流失带来的成本压力。
(6)本发明中,在纯化阶段,采用回流纯化的方式取代了传统的加热、或者加热搅拌、或者加热超声方式,回流纯化方式相较于传统加热、或者加热搅拌纯化方式实现了固液的充分混合,提高了纯化的有效性,相较于加热超声方式产业化实施更为方便,因而更为合理。
附图说明
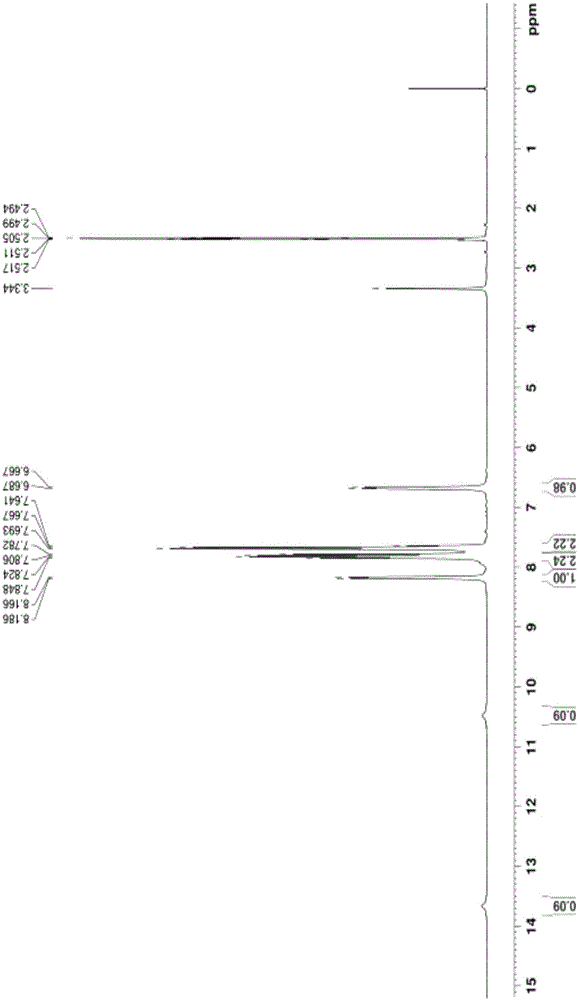
图1为实施例1中首轮制备得到的邻羧基苯甲醛的1h-nmr谱图;
图2为实施例1中首轮制备得到的邻羧基苯甲醛的正离子模式下液相色谱-质谱图;
图3为实施例1中首轮制备得到的邻羧基苯甲醛的负离子模式下液相色谱-质谱图。
具体实施方式
下面通过对本发明进行详细说明,本发明的特点和优点将随着这些说明而变得更为清楚、明确。
在这里专用的词“示例性”意为“用作例子、实施例或说明性”。这里作为“示例性”所说明的任何实施例不必解释为优于或好于其它实施例。尽管在附图中示出了实施例的各种方面,但是除非特别指出,不必按比例绘制附图。
本发明的目的在于提供了一种邻醛基苯基脂肪酸的合成方法,该方法包括:以芳香内酯或者邻甲基苯基脂肪酸为原料,经卤化反应、水解,得到邻醛基苯基脂肪酸。
本发明中,所述芳香内酯的结构为:
其中,x选自氢元素或者卤素,所述卤素选自f、cl或者br,优选为f或者cl,更优选为cl;
n取值0~2。
所述邻甲基苯基脂肪酸的结构为:
在本发明中,卤化反应的过程为:将反应原料、卤源和溶剂加入反应釜中,回流反应,保持反应温度60~100℃,反应时间3~12h,优选反应温度80~90℃,反应时间4~7h,反应得到中间体。
在本发明,溶剂可为溶解反应原料的任意非芳烃类溶剂,优选选自二氯甲烷、三氯甲烷或者四氯甲烷中的任意一种或多种。
在本发明,用于进行卤化反应的卤源选自卤素单质、卤盐或者卤化氢溶液中的一种或多种;
其中,卤素单质选自液溴(br2)或者碘单质(i2);
卤盐为含有卤素离子的盐,选自nabr、kbr、cabr2、nai、ki或者cai2中的一种或多种;优选为nabr或者nai中的一种或多种,更优选为nabr。
卤化氢溶液为含有卤素离子的酸,即卤化氢水溶液,选自hbr或hi。
在本发明中,当以芳香内酯为反应原料进行反应时,若x为h、f或者cl,则进行卤化反应的卤源选自上述卤素单质、卤盐或者卤化氢溶液中的任意一种或多种;
若x为br,则进行卤化反应的卤源选自i2、hi水溶液或者氢碘酸与碱金属氢氧化物反应得到的卤盐,如nai、ki或者cai2中的一种或多种。
由上述卤源的选择可以看出,本发明极大的扩展了工艺过程中传统卤源的选择范围,摈弃了文献报道过的价格昂贵的nbs或者pbr3,很明显,这直接降低了原料的成本。
理论上,经br2或者i2单质进行卤化反应后的上述芳香内酯或者上述邻甲基苯基脂肪酸,经水解反应,即可得到产物邻醛基苯基脂肪酸。本发明人经过研究发现,仅芳香内酯或者邻甲基苯基脂肪酸与卤源进行的简单的卤化—水解反应,不仅卤化反应速率较慢,更为关键的是最终产物的收率并不能得到明显的提高。
另一方面,在采用br2或者i2单质以外的卤源进行反应时,卤化反应速率更慢,这将十分不利于这些卤源的产业化应用。
本发明人经过大量研究,卤化反应过程中加入设定氧化剂,能够解决反应速率慢的问题,这主要基于该设定氧化剂对反应体系中存在的卤素离子的活化作用,即将卤素离子氧化为卤素单质。本发明人还惊奇地发现,由于氧化剂促进了卤源参与反应,氧化剂的加入利于反应原料(芳香内酯、邻甲基苯基脂肪酸)转化率的提高,对此,在产业化合成过程中,能够大幅度降低卤源的用量,利于成本控制、环保以及对溴素、溴化氢等卤源的操作。
在本发明中,卤源为卤素单质、或者卤源为卤盐或卤化氢溶液时,卤化反应过程中氧化剂加入反应体系的时间是不同的,具体地:
(i)当卤源为卤素单质时,在卤素单质耗量介于50%~100%时,加入氧化剂;
(ii)当卤源为卤盐或者卤化氢溶液时,卤源和氧化剂同时或者依次加入反应体系中,以提供活化后的卤源。
优选地,(i’)当卤源为卤素单质时,卤素单质以滴加方式或者分多次加入反应体系中,氧化剂以滴加或者分多次加入反应体系中;
(ii’)当卤源为卤盐或者卤化氢溶液时,卤源一次性加入反应体系中,氧化剂以滴加或者分多次加入反应体系中。
当卤源为卤素单质时,氧化剂不与卤素单质同时加入,原因在于:在卤化反应开始时,反应体系中有充分的能够进行快速反应的卤素单质,不需要加入氧化剂以将卤素单质取代反应原料芳香内酯或邻甲基苯基脂肪酸后产生的卤素离子重新活化为卤素单质,因为此时重新活化产生的卤素单质不会明显促进卤化反应进行;在卤素单质消耗50%~100%时,反应体系中存在较大量的取代反应原料芳香内酯或邻甲基苯基脂肪酸后产生的卤素离子,卤素单质明显减少导致反应速率减慢,此时加入氧化剂,将大量的卤素离子活化(氧化)为卤素单质,重新参与卤化反应,对速率的提升更为有利明显,且氧化剂的有效利用率更高。
同时,我们知道,反应原料芳香内酯或邻甲基苯基脂肪酸均具有苯环结构,存在卤素取代苯环上h元素的可能性,当卤源尤其是卤素单质含量较高(超过2当量的反应原料芳香内酯或邻甲基苯基脂肪酸),发生苯环取代副反应的几率增加明显。氧化剂在卤素单质消耗50%~100%时加入,在控制卤素单质水平满足反应速率要求的前提下,降低了副反应发生。
当卤源为卤盐或者卤化氢溶液时,卤源和氧化剂同时或者依次加入反应体系中,氧化剂即可将卤素离子活化(氧化)为卤素单质,使卤盐或者卤化氢溶液以更高活性的卤素单质形态参与卤化反应,利于反应速率的提高。
在本发明中,(i)当卤源为卤素单质时,卤素单质的用量与反应原料(芳香内酯或者邻甲基苯基脂肪酸)的用量以摩尔比计为(0.55~1.0):1;优选为(0.65~0.80):1。
对于卤素单质而言,理论上1/2当量就足够进行卤化反应,同时生成1当量的卤化氢,实际生产中,我们添加0.55~1.0当量的卤素单质,在耗量50%~100%后,再加氧化剂,以把卤化氢中的卤素置换出来,继续完成反应,以达到减少卤素用量,节省成本的目的。
(ii)当卤源为卤盐或者卤化氢溶液时,卤源的用量与反应原料(芳香内酯或者邻甲基苯基脂肪酸)的用量以摩尔比计为(1.2~2.0):1;优选为(1.5~1.8):1。
在本发明中,氧化剂的目的是将卤源反应过程中生成的或者自身具有的卤素离子活化为卤素单质,因而氧化剂的氧化性必然强于该卤素;所述氧化剂选自氯气(cl2)、双氧水(有效成分h2o2)中的一种或多种,由于双氧水的无毒、无副反应特性,优选为双氧水。
在一种优选地实施方式中,双氧水中过氧化氢的含量为10~28(重量)%。双氧水的浓度对卤化反应速率和最终产物收率有重要影响。双氧水浓度高于28(重量)%,加入反应体系后,与卤素离子迅速发生反应,局部生成高浓度卤素单质,虽然促进反应速率提高,但同时发生苯环取代副反应的机会增加;若双氧水浓度低于10(重量)%,对卤素离子活化为卤素单质的速率降低,进而影响卤化反应整体速率。本发明人发现,双氧水中过氧化氢的含量11~18(重量)%,优选13~15(重量)%时,可以有效平衡反应速率和产物收率两方面。
在一种优选的实施方式中,氧化剂为双氧水,若卤源为卤素单质,氧化剂中过氧化氢的用量与卤素单质的用量以摩尔比计为(0.7~1.2):1,优选为(0.8~1.1):1。当以卤素单质为卤源,1mol卤素单质与原料反应,产生1mol卤素离子,采用(0.7~1.2)mol过氧化氢可有效进行卤素离子的活化。
在另一种优选的实施方式中,氧化剂为双氧水,若卤源为卤盐或者卤化氢溶液,氧化剂中过氧化氢的用量与卤源中卤素离子的用量以摩尔比计为(1.0~1.8):1。当以卤盐或者卤化氢溶液为卤源,1mol卤素离子完全生成卤素单质,需要消耗0.5mol的过氧化氢,但过氧化氢的加入通常不是一次加入,且考虑过氧化氢的无效分解,采用(1.0~1.8)摩尔当量,优选(1.2~1.5)摩尔当量的过氧化氢。
当采用其他氧化剂时,同样满足卤素离子转化为卤素单质的用量需求。
在本发明中,水解的过程为:将卤化反应得到中间体在酸性、中性或碱性条件下,回流,降温,结晶,过滤,得到邻醛基苯基脂肪酸目标产物。
卤化反应得到中间体为3-取代溴化物或3-取代碘化物,能够通过水解形成邻醛基苯基脂肪酸产物。
在本发明中,水解在酸性条件下进行,所用到的酸是无机酸如盐酸、硫酸、磷酸,或有机酸如甲酸、冰醋酸等中的一种或多种。
水解在碱性条件下进行,所用到的碱是无机碱如naoh、koh、ca(oh)2,或有机碱如甲醇钠、乙醇钠等中的一种或多种。
水解在中性条件下进行,采用生产用水即可实现水解。
本发明中,之所以采用回流方式进行水解,而不是传统的加热或者加热搅拌水解的方式的原因在于,本发明人发现,相较于传统的水解方式,回流水解的方式可极大提高水解反应的效率,且采用较少的水解溶剂即可完成整个水解反应。由于回流反应后经降温、结晶、过滤过程后,得到过滤后母液中含有大量卤素离子,以卤盐或者卤化氢溶液形式存在(取决于水解溶剂的酸碱性),我们知道,采用卤盐或者卤化氢溶液可以实施前述的卤化反应,因而过滤后母液有必要回收再次套用至卤化反应中,水解过程中较多的加入水解溶剂,必然造成母液中卤素离子含量过低,不利于母液的直接套用,需要进行浓缩或者补充较多卤素离子后才能直接用于下一轮卤化反应。
同时,本发明中卤盐或卤化氢溶液的选用,其优势不仅仅在于拓展了工艺过程中的溴源,提高溴源的可操作性(溴素或碘单质存储使用相对卤盐或卤化氢溶液相对不便),更为明显的优势在于,水解后母液重新生成了相应的卤盐或卤化氢溶液,解决了产业化合成过程中卤源闭合循环的难题,极大地降低了环保压力,以及卤素流失带来的成本压力。
在一种优选的实施方式中,水解过程中水解溶剂的体积与中间体的质量比为1:(0.7~1.0),优选为1:(0.85~1.0);回流时间为1~3h,优选为1.5~2.5h。
在一种优选的实施方式中,回流反应后降温至0~5℃,恒温结晶6~8h后过滤。
在本发明中,邻醛基苯基脂肪酸的产业化合成方法还包括产物的纯化过程,该纯化过程为:将待纯化产物、活性炭、以及纯化溶剂加入纯化容器中,升温回流,热过滤,搅拌下降温析晶,过滤,冷水洗涤,获得纯化后产物。
在一种优选的实施方式中,所述活性炭与待纯化产物的重量比为0.5%~0.7%。
在一种优选的实施方式中,纯化溶剂可以醇类溶剂或水,优选为水。
在一种优选的实施方式中,回流时间为0.5~1h。
本发明中,采用热过滤方式避免了在低温下纯化后产物以固体形式吸附在活性炭上,降低产品收率。
本发明中,采用回流纯化的方式取代了传统的加热、或者加热搅拌、或者加热超声方式,原因在于,回流纯化方式相较于传统加热、或者加热搅拌纯化方式实现了固液的充分混合,提高了纯化的有效性,相较于加热超声方式产业化实施更为方便,因而更为合理。
在本发明中,回流纯化方式优选实施1至2次,即可实现产品的高度净化。
实施例
以下以邻羧基苯甲醛的合成为例,通过具体实例进一步描述本发明。不过这些实例仅仅是范例性的,并不对本发明的保护范围构成任何限制。
实施例1
250ml四口瓶配置有电磁搅拌、温度计、加料管和冷凝管,加入ccl4100.0ml,邻甲基苯甲酸20.1g(0.15mol),搅拌光照下升温至80℃反应,溴素15.7g(0.10mol)以滴加方式加入四口瓶,回流4小时;然后滴加14%的双氧水30.3g,继续溴化,2小时后,75℃减压脱溶得中间体(3-溴苯酞)33.4g。
100ml四口瓶配置有电磁搅拌、y型管、温度计和冷凝管。加入上步所得中间体33.4g,水35ml。升温回流反应2小时,降温至0℃,8小时后过滤,母液留置待用,滤饼用冰水淋洗,得粗品22.4g。
加入75ml水,0.2g活性炭,回流45min,热过滤,搅拌降温析晶,过滤,冷水洗涤,得18.8g,再用45ml水二次重结晶,得到17.0g(湿),烘干,得到邻羧基苯甲醛14.8g,纯度为99.7%,收率为66.6%。该邻羧基苯甲醛的1h-nmr谱图如图1所示,正离子模式下液相色谱-质谱图如图2所示,负离子模式下液相色谱-质谱图如图3所示。以下示出邻羧基苯甲醛的结构式:
产物结构经1h-nmr分析确定,特征峰:羧基与醛基成环后,形成的羟基上h共振峰(8.16~8.18,d,1h),与羟基相邻碳上h共振峰(6.67~6.68,d,1h),双取代苯环的4个h在苯环区共出现两组共振峰,c位和d位上h的共振峰(7.78~7.85,m,2h),e位和f位上h的共振峰(7.64~7.69,m,2h)。1h-nmr谱图与参考文献(chem.eur.j.2016,22,3009-3018,supportinginformation)中记载一致。
正离子模式下质谱图中,存在m/z=133.0碎片离子峰,该峰为邻羧基苯甲醛(mr=150)断裂掉羟基后形成的。
负离子模式下质谱图中,存在m/z=149.6分子离子峰,该峰与邻羧基苯甲醛负离子模式下形成的分子离子峰一致。
回收套用母液进行下一轮反应:250ml四口瓶配置有电磁搅拌、温度计、加料管和冷凝管。加入ccl4100.0ml,加入上述水解母液,补加溴素1.6g(0.01mol),邻甲基苯甲酸20.1g(0.15mol),重复上述步骤,最终得到目标产物14.5g,纯度为99.8%,收率为65.3%。
实施例2
250ml四口瓶配置有电磁搅拌、温度计、加料管和冷凝管,加入ccl4100.0ml,苯酞20.1g(0.15mol),搅拌光照下升温至80℃反应,溴素15.7g(0.10mol)以滴加方式加入四口瓶,回流4小时;然后滴加28%的双氧水15.15g,继续溴化,2小时后,75℃减压脱溶得中间体(3-溴苯酞)32.0g。
100ml四口瓶配置有电磁搅拌、y型管、温度计和冷凝管。加入上步所得中间体32.0g,水35ml。升温回流反应2小时,降温至5℃,8小时后过滤,母液留置待用,滤饼用冰水淋洗,得粗品21.4g。
加入75ml水,0.2g活性炭,回流45min,热过滤,搅拌降温析晶,过滤,冷水洗,得18.6g,再用45ml水二次重结晶,得到17.0g(湿),烘干,得到邻羧基苯甲醛14.3g,纯度为99.9%,收率为63.5%。
回收套用母液进行下一轮反应:250ml四口瓶配置有电磁搅拌、温度计和冷凝管。加入ccl4100.0ml,加入上述水解母液,补加溴素1.6g(0.01mol),苯酞20.1g(0.15mol),重复上述步骤,最终得到目标产物14.0g,纯度为99.7%,收率为62.2%。
实施例3
250ml四口瓶配置有电磁搅拌、温度计、加料管和冷凝管,加入ccl4100.0ml,nabr水溶液(23.2gnabr溶于100ml水,0.225mol),邻羧基甲苯20.1g(0.15mol),搅拌光照下升温至80℃反应,回流6小时,回流过程中滴加14%的双氧水71g;溴化完成后,75℃减压脱溶得中间体32.8g。
100ml四口瓶配置有电磁搅拌、y型管、温度计和冷凝管。加入上步所得中间体32.8g,水35ml。升温回流反应2小时,降温至0℃,8小时后过滤,母液留置待用,滤饼用冰水淋洗,得粗品22.8g。
加入75ml水,0.2g活性炭,回流45min,热过滤,搅拌降温析晶,过滤,冷水洗涤,得18.0g,再用45ml水二次重结晶,得到16.5g(湿),烘干,得到邻羧基苯甲醛14.3g,纯度为99.9%,收率为64.4%。
回收套用母液进行下一轮反应:250ml四口瓶配置有电磁搅拌、温度计、加料管和冷凝管。加入ccl4100.0ml,加入上述水解母液,补加nabr2.06g(0.02mol),邻羧基甲苯20.1g(0.15mol),重复上述步骤,最终得到目标产物14.2g,纯度为99.8%,收率为63.9%。
实施例4
250ml四口瓶配置有电磁搅拌、温度计、加料管和冷凝管,加入ccl4100.0ml,nabr水溶液(23.2gnabr溶于100ml水,0.225mol),3-氯苯酞25.4g(0.15mol),搅拌光照下升温至80℃反应,回流6小时,回流过程中滴加14%的双氧水71g;溴化完成后,75℃减压脱溶得中间体(3-溴苯酞)32.6g。
100ml单口瓶加电磁搅拌、y型管、温度计和冷凝管。加入上步所得中间体32.6g,水35ml。升温回流反应2小时,降温至0℃,8小时后过滤,母液留置待用,滤饼用冰水淋洗,得粗品23.8g。
加入75ml水,0.2g活性炭,回流45min,热过滤,搅拌降温析晶,过滤,冷水洗,得19.2g,再用45ml水二次重结晶,得到17.5g(湿),烘干,得到邻羧基苯甲醛15.3g,纯度为99.8%,收率为67.9%。
回收套用母液进行下一轮反应:250ml四口瓶配置有电磁搅拌、温度计、加料管和冷凝管。加入ccl4100.0ml,加入上述水解母液,补加nabr2.06g(0.02mol),3-氯苯酞25.4g(0.15mol),重复上述步骤,最终得到目标产物15.6g,纯度为99.8%,收率为69.3%。
实施例5
反应条件与实施例1相同,区别仅在于:首次反应过程中,溴素的用量为0.12mol。
实施例6
反应条件与实施例1相同,区别仅在于:首次反应过程中,溴素一次性加入反应容器中。
实施例7
反应条件与实施例3相同,区别仅在于:首次反应过程中,nabr的用量为0.18mol。
实施例8
反应条件与实施例3相同,区别仅在于:首次反应过程中,nabr的用量为0.30mol。
实施例9
反应条件与实施例3相同,区别仅在于:首次反应过程中,采用28%的双氧水35.5g。
实施例10
反应条件与实施例3相同,区别仅在于:首次反应过程中,采用40%的双氧水24.8g。
实施例11
反应条件与实施例3相同,区别仅在于:首次反应过程中,采用8%的双氧水124g。
对比例
对比例1
反应条件与实施例1相同,区别仅在于:首次反应过程中,溴素的用量为0.25mol。
对比例2
反应条件与实施例3相同,区别仅在于:首次反应过程中,纯化过程中采用加热搅拌方式替代回流纯化方式。
实施例1~11和对比例1~2的反应结果汇总见表1:
表1
以上结合具体实施方式和范例性实例对本发明进行了详细说明,不过这些说明并不能理解为对本发明的限制。本领域技术人员理解,在不偏离本发明精神和范围的情况下,可以对本发明技术方案及其实施方式进行多种等价替换、修饰或改进,这些均落入本发明的范围内。本发明的保护范围以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!