一种含柔性硅氧链节的芳香胺官能化碳硼烷化合物及其制备方法与应用与流程
本发明属于高分子材料技术领域,具体涉及一种含柔性硅氧链节的芳香胺官能化碳硼烷化合物及其制备方法与应用。
背景技术:
航天航空等高技术领域的不断发展,对聚氨酯、聚酰亚胺、聚酰胺、聚脲、环氧树脂、聚硅氧烷等一系列的高分子材料的使用温度提出了越来越苛刻的需求,这就需要不断提高现有的有机高分子材料的热降解温度。从分子结构设计上引入特殊的结构成为制备先进高分子材料的途径之一。以含硅、硼、氮等无机元素组成的特殊结构受到青睐,比如呈六面体结构的倍半硅氧烷、二十面体的碳硼烷结构等。尤其是作为二十面体结构的碳硼烷,以其高含硼的立体结构在耐高温、抗原子氧、抗中子辐照等领域表现出潜在的应用,比如专利文献(申请号201610544400.8)提出了一种采用苯基亚甲基过渡的胺基结构,并给出了含该结构的碳硼烷的制备方法以及用其制备的聚酰亚胺在中子防护领域的潜在应用;专利文献(申请号201510447823.3)给出了使用含芳香胺结构的碳硼烷制备聚酰亚胺的方法,提出其在耐高温方面的应用。
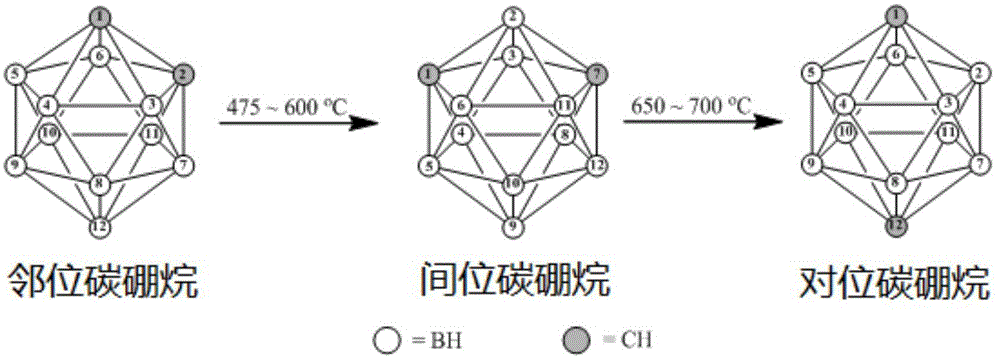
碳硼烷是具有邻位(o-carborane)、间位(m-carborane)、对位(p-carborane)三种异构体的大体积、高含硼量的立体结构,其结构如图1所示。然而,这种特殊结构的碳硼烷是一类易升华的固体,若想在分子水平上将其引入聚合物的结构中,需要对碳硼烷的结构进行修饰,实现碳硼烷的官能化。因此,如何制备耐高温的有机官能团修饰的笼型碳硼烷结构,可以使得高分子材料的耐高温、抗氧化、抗中子辐照等综合性能都大幅度的提高,成为碳硼烷改性高分子材料的关键技术挑战。
技术实现要素:
本发明主要提出了一种含柔性硅氧链节的芳香胺官能化碳硼烷化合物及其制备方法与应用,目的是提供一种分子水平上的有机/无机杂化结构。本发明在碳硼烷的笼型结构上修饰含柔性硅氧链节的芳香胺基官能团,该结构创新性的将柔性硅氧链节与大体积、高含硼量的碳硼烷立体结构结合在一起,并通过活性的芳香胺赋予该结构高反应活性,可以方便的将两种结构在分子水平上引入有机高分子聚合物中,制备得到的有机高分子聚合物具有突出的耐高温特性。
本发明的目的通过以下技术方案实现:
一种含柔性硅氧链节的芳香胺官能化碳硼烷化合物,其结构如下式(a)所示,
其中,“-cb10h10c-”代表邻位碳硼烷基、间位碳硼烷基或对位碳硼烷基;
r为-芳基-、-芳基-芳基-、-芳基-芳基-芳基-、或任选被一个或多个c1-10烷基或芳基取代的-芳基-、-芳基-芳基-、-芳基-芳基-芳基-;
r1、r2、r3、r4相同或不同,彼此独立地选自h、无取代或任选被一个或多个ra取代的c1-10烷基、c3-20环烷基、芳基;
每一个ra相同或不同,彼此独立地选自卤素、cn、c1-10烷基、c1-10烷氧基或c3-20环烷基;
根据本发明的实施方案,r选自苯基、甲苯基、乙基苯基、联苯基、萘基、蒽基、三联苯基。
根据本发明的实施方案,r1、r2、r3、r4相同或不同,彼此独立地选自无取代或取代的h、c1-6烷基、芳基,例如甲基、乙基、三氟丙基、腈丙基、苯基、甲苯基、萘基;
m选自0-10的整数,优选0-5,例如0、1、2、3。
本发明还提供上述式(a)所示化合物的制备方法,包括如下步骤:
(1)碳硼烷(i)与金属类去质子化试剂反应,得到碳硼烷活性中间体(ii);
(2)碳硼烷活性中间体(ii)与卤代硅烷或卤代硅氧烷反应,得到化合物(iii);
(3)化合物(iii)进行水解反应,得到化合物(iv);
(4)将化合物(iv)与金属类去质子化试剂反应,得到化合物(v);
(5)将化合物(v)与卤代芳香烃化合物rx反应,得到化合物(vi);
(6)化合物(vi)进行硝化反应,得到化合物(vii);
(7)化合物(vii)进行还原反应,得到化合物(a)。
其中,“-cb10h10c-”、r1、r2、r3、r4、r、m具有上文所述的定义;m选自金属,优选为碱金属,例如li、na、k;x1选自cl、br或i,x为卤素,例如cl、br或i。
根据本发明,所述碳硼烷选自邻位碳硼烷、间位碳硼烷或对位碳硼烷中的一种、两种或三种的混合物;
根据本发明,所述金属类去质子化试剂选自含元素m的化合物,例如选自正丁基锂、仲丁基锂、叔丁基锂、六甲基二硅基胺基锂、二异丙基胺基锂、乙炔钠、双(三甲基硅基)氨基钠、氢化钠、氢化钾、双(三甲基硅基)氨基钾;
根据本发明,所述卤代硅烷或卤代硅氧烷的结构如下式(viii)所示;其中,m、x1、r1、r2、r3、r4具有上文所述的定义;x1、x2相同或不同,彼此独立地选自cl、br或i;
根据本发明,步骤(1)中,
所述反应可以在溶剂中进行,所述溶剂可以为醚类溶剂,例如乙醚、四氢呋喃、乙二醇二甲醚、二乙二醇二甲醚中的一种、两种或多种的混合物;
所述碳硼烷(i)与金属类去质子化试剂的物质的量的比例为1:(1.50-2.50),优选为1:(2.05-2.10),例如1:2.05、1:2.08、1:2.10;
所述反应可以在惰性气体保护下进行,例如氮气保护;
所述反应的温度可以为20-30℃,例如室温;
所述反应的时间可以为0.2-4小时,优选为0.5-3小时,例如0.5小时、2小时、3小时;
根据本发明,步骤(2)中,
所述碳硼烷(i)与卤代硅烷或卤代硅氧烷的物质的量的比例为1:(1.50-2.50),优选为1:(2.05-2.10),例如1:2.05、1:2.08、1:2.10;
所述反应可以在加热条件下进行,例如在回流条件下进行;
所述反应的时间可以为1-27小时,优选为4-24小时,例如4小时、12小时、24小时;
根据本发明,步骤(3)中,
所述反应可以加入水;
所述碳硼烷(i)与水的物质的量的比例为1:(1.50-2.50),优选为1:(2.05-2.10),例如1:2.05、1:2.08、1:2.10;
所述反应中还可以加入有机溶剂,所述有机溶剂可以为醚类溶剂,例如乙醚、四氢呋喃、乙二醇二甲醚;
所述水与有机溶剂的体积比可以为(1-14):100,优选为(5-10):100,例如10:100、5:100、8:100;
所述反应可以加入酸吸收剂,所述酸吸收剂可以为碱性化合物,例如氨水、吡啶、碳酸氢钠;所述酸吸收剂可以吸附水解过程中产生的卤化氢;
所述碳硼烷(i)与酸吸收剂的物质的量的比例为1:(1.50-2.50),优选为1:(2.05-2.10),例如1:2.05、1:2.08、1:2.10;
所述反应的温度可以为(-35℃)~0℃,优选为(-30℃)~(-5℃),例如-30℃、-5℃、-20℃;
所述反应的时间可以为0.1-1.4小时,例如0.5-1小时,例如0.5小时、1小时;
根据本发明,步骤(4)中,
所述反应可以在溶剂中进行,所述溶剂可以为醚类溶剂,例如乙醚、四氢呋喃、乙二醇二甲醚;
所述化合物(iv)与金属类去质子化试剂的物质的量的比例为1:(1.50-2.50),优选为1:(2.05-2.10),例如1:2.05、1:2.08、1:2.10;
所述反应可以在惰性气体保护下进行,例如氮气保护;
所述反应的温度可以为20-30℃,例如室温;
所述反应的时间可以为0.2-4小时,优选为0.5-3小时,例如0.5小时、2小时、3小时;
根据本发明,步骤(5)中,
所述化合物(iv)与卤代芳香烃化合物rx的物质的量的比例为1:(1.50-2.50),优选为1:(2.05-2.10),例如1:2.05、1:2.08、1:2.10;
所述反应可以在加热条件下进行,例如在回流条件下进行;
所述反应的时间可以为1-27小时,优选为4-24小时,例如4小时、12小时、24小时;
根据本发明,步骤(6)中,
所述反应中可以加入溶剂,所述溶剂可以选自卤代烃类溶剂、芳烃类溶剂、烷烃类溶剂,例如二氯甲烷、苯、甲苯、正己烷、环己烷等的一种或几种的混合物;
所述反应可以加入硫酸和硝酸的混合物,优选为浓硫酸和浓硝酸的混合溶液;
所述硫酸与硝酸的体积比可以为1:(1-3),优选为1:2;
所述化合物(vi)与硝酸的物质的量的比例为1:(1.55-2.66),优选为1:(2.05-2.16),例如1:2.05、1:2.10、1:2.16;
所述反应的温度可以为20-30℃,例如室温;
所述反应的时间可以为1-11小时,优选为4-8小时,例如4小时、6小时、8小时;
根据本发明,步骤(7)中,
所述反应中可以加入溶剂,所述溶剂选自醚类溶剂,例如乙醚、四氢呋喃、乙二醇二甲醚、二乙二醇二甲醚等的一种或几种的混合物;
所述反应可以加入还原剂,所述还原剂可以为铁/盐酸、水合肼等;
所述化合物(vii)与还原剂的物质的量的比例为1:(1-7),优选为1:(2-6),例如1:2、1:3、1:6;
所述反应可以在加热条件下进行,所述反应的温度可以为50-125℃,优选为65-110℃,例如65℃、85℃、110℃;
所述反应的时间可以为0.5-6.5小时,优选为1-6小时,例如1小时、4小时、6小时;
本发明还提供上述式(a)化合物的用途,其可以用作聚合物的交联剂。
根据本发明,所述式(a)化合物作为交联剂与聚合物进行交联反应,所述聚合物可以为能够与式(a)化合物中胺基进行反应的聚合物体系,例如具有环氧基、羟基等的聚合物,具体的聚合物可为:环氧树脂、有机硅树脂、有机硅橡胶。
本发明还提供上述式(a)化合物的又一用途,其可以用作聚合物单体。
根据本发明,所述式(a)化合物作为单体与其他单体进行聚合反应,所述式(a)化合物可以作为单体来制备含有n元素组成的官能团的聚合物,例如其可以用于制备聚酰亚胺、聚酰胺、聚脲、聚硅氮烷等聚合物。
一种聚合物,其包括如下所示的式(b)或式(c)结构;
式中,“-cb10h10c-”、r1、r2、r3、r4、r、m具有上文所述的定义。
根据本发明,所述聚合物为交联聚合物,所述式(b)所示结构为交联聚合物中的交联单元;即所述式(b)所示结构是通过交联剂引入到聚合物中。所述进行交联的聚合物例如具有环氧基、羟基等的聚合物,具体的聚合物可为:环氧树脂、有机硅树脂、有机硅橡胶。
根据本发明,所述聚合物中,所述式(b)或式(c)所示结构为聚合物中的重复单元;
根据本发明,所述聚合物中还可以包括以下的重复单元中的一种或两种:
根据本发明,所述聚合物的分子量例如为:1000~1000000。
本发明还提供上述聚合物的制备方法,所述制备方法包括以下方法的任一种:
(1)式(a)化合物作为交联剂:将需要交联固化的聚合物体系与本发明所述式(a)化合物进行反应,得到聚合物;
或
(2)式(a)化合物作为单体:将本发明所述式(a)化合物作为聚合单体与其他聚合单体进行反应,得到聚合物;
根据本发明,方法(1)中,需要交联固化的聚合物体系可以为具有可与胺基反应的基团的聚合物体系,优选的具有环氧基、羟基等的聚合物,例如聚合物可为:环氧树脂、有机硅树脂、有机硅橡胶。
根据本发明,方法(1)中,需要固化的聚合物体系与式(a)化合物的质量比为100:(5~100)。
根据本发明,方法(1)中,所述反应还可以任选地加入其他交联剂。例如本领域中常用的交联剂。所述常用的交联剂包括但不限于脂肪胺、芳香胺、聚硅氮烷、聚酰胺、氨基树脂、含氨基硅氧烷中的一种、两种或多种。
所述其他交联剂与式(a)化合物的质量比是任意的。
根据本发明,方法(2)中,所述其他聚合单体为具有可与氨基进行反应的基团的化合物,所述其他聚合物单体可为:
优选地,所述式(a)化合物与其他聚合单体的质量比是任意的。
本发明还提供上述聚合物的应用,其可以用于制备纤维增强复合材料、防护涂层、粘接密封材料等。
有益效果
本发明提供了一种含柔性硅氧链节的芳香胺官能化碳硼烷化合物及其制备方法与应用。本发明的制备方法简单,操作方便,适于工业化的大规模生产。而且,本发明化合物可以用作交联剂或聚合物的单体引入聚合物中,从而得到一类有机/无机杂化组元的聚合物。所述聚合物因引入柔性硅氧链节赋予本征的韧性,使得聚合物的韧性提高,同时引入立体、富硼的碳硼烷结构赋予聚合物耐更高温度、抗氧化性能提高、极限氧指数和水平垂直燃烧等本征阻燃性能提高、抗中子辐照能力增强、具有更高温度和更大气流冲蚀下的烧蚀性能、同时能提高空间环境下的抗原子氧侵蚀等优良性能,其可用作纤维增强复合材料、防护涂层、粘接密封材料的树脂基体。
术语定义和说明
除非另有说明,本申请说明书和权利要求书中记载的基团和术语定义,包括其作为实例的定义、示例性的定义、优选的定义、表格中记载的定义、实施例中具体化合物的定义等,可以彼此之间任意组合和结合。这样的组合和结合后的基团定义及化合物结构,应当属于本申请说明书记载的范围内。
术语“c1-10烷基”应理解为优选表示具有1~10个碳原子的直链或支链饱和一价烃基,优选为c1-6烷基。“c1-6烷基”应理解为优选表示具有1、2、3、4、5或6个碳原子的直链或支链饱和一价烃基。所述烷基是例如甲基、乙基、丙基、丁基、戊基、己基、异丙基、异丁基、仲丁基、叔丁基、异戊基、2-甲基丁基、1-甲基丁基等。
术语“c3-20环烷基”应理解为表示饱和的一价单环或双环烃环,其具有3~20个碳原子,优选“c3-10环烷基”。术语“c3-10环烷基”应理解为表示饱和的一价单环或双环烃环,其具有3、4、5、6、7、8、9或10个碳原子。所述c3-10环烷基可以是单环烃基,如环丙基、环丁基、环戊基、环己基、环庚基、环辛基、环壬基或环癸基,或者是双环烃基如十氢化萘环。
术语“芳基”应理解为具有6-20个(优选6-14个)碳原子的单环或多环芳香族基团,代表性的芳基包括:苯基、萘基、蒽基、芘基等。
附图说明
图1为三种碳硼烷异构体的结构图。
图2为实施例1中化合物(a-1)的1h-nmr谱图。
图3为实施例1中化合物(a-1)的13c-nmr谱图。
图4为实施例1中化合物(a-1)的11b-nmr谱图。
图5为实施例1中化合物(a-1)的29si-nmr谱图。
图6为实施例2中化合物(a-2)的1h-nmr谱图。
图7为实施例2中化合物(a-2)的13c-nmr谱图。
图8为实施例2中化合物(a-2)的11b-nmr谱图。
图9为实施例2中化合物(a-2)的29si-nmr谱图。
图10为实施例3中化合物(a-3)的1h-nmr谱图。
图11为实施例3中化合物(a-3)的13c-nmr谱图。
图12为实施例3中化合物(a-3)的11b-nmr谱图。
图13为实施例3中化合物(a-3)的29si-nmr谱图。
具体实施方式
下文将结合具体实施例对本发明做更进一步的详细说明。应当理解,下列实施例仅为示例性地说明和解释本发明,而不应被解释为对本发明保护范围的限制。凡基于本发明上述内容所实现的技术均涵盖在本发明旨在保护的范围内。
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
实施例1
步骤(1):由邻位碳硼烷(i-1)制备邻位碳硼烷的二锂盐(ii-1)活性中间体
将由高低温一体化温度控制系统、强力搅拌系统、冷凝回流系统、真空系统、反应釜组成的反应装置经抽真空、氮气置换的方式除去整套反应装置中的空气后,在氮气保护下,将1mol(144.2g)的邻位碳硼烷(i-1)、2l经除水干燥的乙醚加入到反应釜中,经搅拌溶解;使用高低温一体化温度控制系统的低温制冷功能将该邻位碳硼烷(i-1)的乙醚溶液冷却到-60℃;将含叔丁基锂为2.10mol的叔丁基锂溶液使用加料泵在氮气保护下缓慢的滴加到反应釜中;在叔丁基锂溶液滴加完毕后,关闭高低温一体化温度控制系统的低温制冷功能,使得反应装置的温度缓慢的升高到室温后继续搅拌反应3h得到活性中间体—邻位碳硼烷的二锂盐(ii-1)。
步骤(2):由制备的邻位碳硼烷的二锂盐(ii-1)活性中间体制备双(氯代二甲基硅基)邻位碳硼烷(iii-1)
通过液体计量装置缓慢的将2.05mol的二甲基二氯硅烷滴加到步骤(1)的反应装置中,滴加完毕后,打开反应装置所连接的高低温一体化温度控制系统进行缓慢升温至34℃,并控制在该温度下回流、搅拌反应24h得到双(氯代二甲基硅基)邻位碳硼烷(iii-1)。
步骤(3):由制备的双(氯代二甲基硅基)邻位碳硼烷(iii-1)制备双(羟基二甲基硅基)邻位碳硼烷(iv-1)
将水稀释在乙醚溶剂中,控制两者的体积比为10:100;打开步骤(1)的反应装置所连接的高低温一体化温度控制系统将反应体系的温度控制在-5℃,通过液体计量装置缓慢的将含2.05mol水的乙醚溶液滴加到步骤(1)的反应装置中,并向反应体系中加入2.05mol的氨水作为酸吸收剂,控制反应体系在-5℃的低温下搅拌反应0.5h,得到双(羟基二甲基硅基)邻位碳硼烷(iv-1)。经过过滤、蒸馏后使用正己烷重结晶后得到双(羟基二甲基硅基)邻位碳硼烷(iv-1)产物。
步骤(4):由制备的双(羟基二甲基硅基)邻位碳硼烷(iv-1)制备双(锂氧基二甲基硅基)邻位碳硼烷(v-1)
将由高低温一体化温度控制系统、强力搅拌系统、冷凝回流系统、真空系统、反应釜组成的反应装置经抽真空、氮气置换的方式除去整套反应装置中的空气后,在氮气保护下,将1mol的双(羟基二甲基硅基)邻位碳硼烷(iv-1)、2l经除水干燥的乙醚加入到反应釜中,经搅拌溶解;使用高低温一体化温度控制系统的低温制冷功能将该双(羟基二甲基硅基)邻位碳硼烷(iv-1)的乙醚溶液冷却到-60℃;将含叔丁基锂为2.10mol的叔丁基锂溶液使用加料泵在氮气保护下缓慢的滴加到反应釜中;在叔丁基锂溶液滴加完毕后,关闭高低温一体化温度控制系统的低温制冷功能,使得反应装置的温度缓慢的升高到室温后继续搅拌反应3h得到活性中间体—双(锂氧基二甲基硅基)邻位碳硼烷(v-1)。
步骤(5):由制备的双(锂氧基二甲基硅基)邻位碳硼烷(v-1)制备双(苯氧基二甲基硅基)邻位碳硼烷(vi-1)
通过液体计量装置缓慢的将2.05mol的碘苯滴加到步骤(4)的反应装置中,滴加完毕后,打开反应装置所连接的高低温一体化温度控制系统进行缓慢升温至34℃,并控制在该温度下回流、搅拌反应24h得到双(苯氧基二甲基硅基)邻位碳硼烷(vi-1)。经过过滤、蒸馏后使用正己烷重结晶后得到双(苯氧基二甲基硅基)邻位碳硼烷(vi-1)产物。
步骤(6):由制备的双(苯氧基二甲基硅基)邻位碳硼烷(vi-1)制备双((硝基苯)-氧基二甲基硅基)邻位碳硼烷(vii-1)
将1mol双(苯氧基二甲基硅基)邻位碳硼烷(vi-1)加入由高低温一体化温度控制系统、强力搅拌系统、冷凝回流系统、真空系统、反应釜组成的反应装置中,开动搅拌,加入2l二氯甲烷溶解;开动高低温一体化温度控制系统的制冷,将反应装置的温度冷却到0℃后,缓慢的滴加浓硫酸和浓硝酸的混合溶液(浓硫酸和浓硝酸的体积比为1:2),控制所加入到混合酸液中的硝酸的物质的量为2.05mol,滴加完成后关闭高低温一体化温度控制系统的制冷,使得反应体系的温度缓慢的升到室温,在室温下搅拌反应4h后,得到含双((硝基苯)-氧基二甲基硅基)邻位碳硼烷(vii-1)的产物。将反应产物缓慢的加入2l冰水混合物中,再使用乙醚萃取,然后用饱和碳酸氢钠溶液洗涤有机相至中性,将有机相使用无水硫酸钠干燥后蒸馏,得到双((硝基苯)-氧基二甲基硅基)邻位碳硼烷(vii-1)。
步骤(7):由制备的双((硝基苯)-氧基二甲基硅基)邻位碳硼烷(vii-1)制备双((氨基苯)-氧基二甲基硅基)邻位碳硼烷(a-1)
将1mol双((硝基苯)-氧基二甲基硅基)邻位碳硼烷(vii-1)加入由加热控制系统、强力搅拌系统、冷凝回流系统、真空系统、反应釜组成的反应装置中,开动搅拌,加入2l四氢呋喃溶解;加入3mol铁/盐酸还原体系后开动加热控制系统,将反应装置的温度升高到65℃,搅拌反应4h后,得到双((氨基苯)-氧基二甲基硅基)邻位碳硼烷(a-1)的产物。向反应产物中滴入稀盐酸调节ph小于1,加入水2l后使用二氯甲烷萃取,向水相中加入碳酸钠调节ph>10,并用二氯甲烷萃取后合并有机相,然后使用无水硫酸镁干燥后蒸馏,得到双((氨基苯)-氧基二甲基硅基)邻位碳硼烷(a-1)。
化合物(a-1)的1h-nmr谱图、13c-nmr谱图、11b-nmr谱图、29si-nmr谱图见图2-5。
实施例2
步骤(1):由间位碳硼烷(i-2)制备间位碳硼烷的二锂盐(ii-2)活性中间体
将由高低温一体化温度控制系统、强力搅拌系统、冷凝回流系统、真空系统、反应釜组成的反应装置经抽真空、氮气置换的方式除去整套反应装置中的空气后,在氮气保护下,将1.5mol(216.3g)的间位碳硼烷(i-2)、5l经除水干燥的四氢呋喃加入到反应釜中,经搅拌溶解;使用高低温一体化温度控制系统的低温制冷功能将该间位碳硼烷(i-2)的四氢呋喃溶液冷却到-40℃;将含正丁基锂为3.12mol的正丁基锂溶液使用加料泵在氮气保护下缓慢的滴加到反应釜中;在正丁基锂溶液滴加完毕后,关闭高低温一体化温度控制系统的低温制冷功能,使得反应装置的温度缓慢的升高到室温后继续搅拌反应2h得到活性中间体—间位碳硼烷的二锂盐(ii-2)。
步骤(2):由制备的间位碳硼烷的二锂盐(ii-2)活性中间体制备双(3-溴代-1,1,3,3-四苯基二硅氧基)间位碳硼烷(iii-2)
通过液体计量装置缓慢的将3.15mol的1,3-二溴代-1,1,3,3-四苯基二硅氧烷滴加到步骤(1)的反应装置中,滴加完毕后,打开反应装置所连接的高低温一体化温度控制系统进行缓慢升温至65℃,并控制在该温度下回流、搅拌反应12h得到双(3-溴代-1,1,3,3-四苯基二硅氧基)间位碳硼烷(iii-2)。
步骤(3):由制备的双(3-溴代-1,1,3,3-四苯基二硅氧基)间位碳硼烷(iii-2)制备双(3-羟基-1,1,3,3-四苯基二硅氧基)间位碳硼烷(iv-2)
将水稀释在四氢呋喃溶剂中,控制两者的体积比为5:100;打开步骤(1)的反应装置所连接的高低温一体化温度控制系统将反应体系的温度控制在-30℃,通过液体计量装置缓慢的将含3.15mol水的四氢呋喃溶液滴加到步骤(1)的反应装置中,并向反应体系中加入3.15mol的碳酸氢钠作为酸吸收剂,控制反应体系在-30℃的低温下搅拌反应1h,得到双(3-羟基-1,1,3,3-四苯基二硅氧基)间位碳硼烷(iv-2)。经过过滤、蒸馏后使用甲苯重结晶后得到双(3-羟基-1,1,3,3-四苯基二硅氧基)间位碳硼烷(iv-2)。
步骤(4):由制备的双(3-羟基-1,1,3,3-四苯基二硅氧基)间位碳硼烷(iv-2)制备双(3-锂氧-1,1,3,3-四苯基二硅氧基)间位碳硼烷(v-2)
将由高低温一体化温度控制系统、强力搅拌系统、冷凝回流系统、真空系统、反应釜组成的反应装置经抽真空、氮气置换的方式除去整套反应装置中的空气后,在氮气保护下,将1.5mol的双(3-羟基-1,1,3,3-四苯基二硅氧基)间位碳硼烷(iv-2)、5l经除水干燥的四氢呋喃加入到反应釜中,经搅拌溶解;使用高低温一体化温度控制系统的低温制冷功能将该双(3-羟基-1,1,3,3-四苯基二硅氧基)间位碳硼烷(iv-2)的四氢呋喃溶液冷却到-40℃;将含正丁基锂为3.12mol的正丁基锂溶液使用加料泵在氮气保护下缓慢的滴加到反应釜中;在正丁基锂溶液滴加完毕后,关闭高低温一体化温度控制系统的低温制冷功能,使得反应装置的温度缓慢的升高到室温后继续搅拌反应2h得到活性中间体—双(3-锂氧-1,1,3,3-四苯基二硅氧基)间位碳硼烷(v-2)。
步骤(5):由制备的双(3-锂氧-1,1,3,3-四苯基二硅氧基)间位碳硼烷(v-2)制备双(3-苄氧-1,1,3,3-四苯基二硅氧基)间位碳硼烷(vi-2)
通过液体计量装置缓慢的将3.15mol的碘代甲苯滴加到步骤(4)的反应装置中,滴加完毕后,打开反应装置所连接的高低温一体化温度控制系统进行缓慢升温至65℃,并控制在该温度下回流、搅拌反应12h得到双(3-苄氧-1,1,3,3-四苯基二硅氧基)间位碳硼烷(vi-2)。经过过滤、蒸馏后使用甲苯重结晶后得到双(3-苄氧-1,1,3,3-四苯基二硅氧基)间位碳硼烷(vi-2)。
步骤(6):由制备的双(3-苄氧-1,1,3,3-四苯基二硅氧基)间位碳硼烷(vi-2)制备双(3-(硝基苄)-氧-1,1,3,3-四苯基二硅氧基)间位碳硼烷(vii-2)
将2mol双(3-苄氧-1,1,3,3-四苯基二硅氧基)间位碳硼烷(vi-2)加入由高低温一体化温度控制系统、强力搅拌系统、冷凝回流系统、真空系统、反应釜组成的反应装置中,开动搅拌,加入5l甲苯溶解;开动高低温一体化温度控制系统的制冷,将反应装置的温度冷却到-20℃后,缓慢的滴加浓硫酸和浓硝酸的混合溶液(浓硫酸和浓硝酸的体积比为1:2),控制所加入到混合酸液中的硝酸的物质的量为4.2mol,滴加完成后关闭高低温一体化温度控制系统的制冷,使得反应体系的温度缓慢的升到室温,在室温下搅拌反应8h后,得到含双(3-(硝基苄)-氧-1,1,3,3-四苯基二硅氧基)间位碳硼烷(vii-2)的产物。将反应产物缓慢的加入5l冰水混合物中,再使用石油醚萃取,然后用饱和碳酸氢钾溶液洗涤有机相至中性,将有机相使用无水硫酸镁干燥后蒸馏,得到双(3-(硝基苄)-氧-1,1,3,3-四苯基二硅氧基)间位碳硼烷(vii-2)。
步骤(7):由制备的双(3-(硝基苄)-氧-1,1,3,3-四苯基二硅氧基)间位碳硼烷(vii-2)制备双(3-(氨基苄)-氧-1,1,3,3-四苯基二硅氧基)间位碳硼烷(a-2)
将2mol双(3-(硝基苄)-氧-1,1,3,3-四苯基二硅氧基)间位碳硼烷(vii-2)加入由加热控制系统、强力搅拌系统、冷凝回流系统、真空系统、反应釜组成的反应装置中,开动搅拌,加入5l乙二醇二甲醚溶解;加入4mol铁/盐酸还原体系后开动加热控制系统,将反应装置的温度升高到85℃,搅拌反应1h后,得到双(3-(氨基苄)-氧-1,1,3,3-四苯基二硅氧基)间位碳硼烷(a-2)的产物。向反应产物中滴入稀盐酸调节ph小于1,加入水5l后使用甲苯萃取,向水相中加入碳酸钠调节ph>10,并用甲苯萃取后合并有机相,然后使用无水硫酸镁干燥后蒸馏,得到双(3-(氨基苄)-氧-1,1,3,3-四苯基二硅氧基)间位碳硼烷(a-2)。
化合物(a-2)的1h-nmr谱图、13c-nmr谱图、11b-nmr谱图、29si-nmr谱图见图6-9。
实施例3
步骤(1):由对位碳硼烷(i-3)制备对位碳硼烷的二钠盐(ii-3)活性中间体
将由高低温一体化温度控制系统、强力搅拌系统、冷凝回流系统、真空系统、反应釜组成的反应装置经抽真空、氮气置换的方式除去整套反应装置中的空气后,在氮气保护下,将0.5mol(72.1g)的对位碳硼烷(i-3)、1l经除水干燥的乙二醇二甲醚加入到反应釜中,经搅拌溶解;使用高低温一体化温度控制系统的低温制冷功能将该对位碳硼烷(i-3)的乙二醇二甲醚溶液冷却到-10℃;将含乙炔钠为1.025mol的乙炔钠溶液使用加料泵在氮气保护下缓慢的滴加到反应釜中;在乙炔钠溶液滴加完毕后,关闭高低温一体化温度控制系统的低温制冷功能,使得反应装置的温度缓慢的升高到室温后继续搅拌反应0.5h得到活性中间体—对位碳硼烷的二钠盐(ii-3)。
步骤(2):由制备的对位碳硼烷的二钠盐(ii-3)活性中间体制备双(5-氯代-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(iii-3)
通过液体计量装置缓慢的将1.04mol的1,5-二氯代-1,3,5-三甲基-1,3,5-三苯基三硅氧烷滴加到步骤(1)的反应装置中,滴加完毕后,打开反应装置所连接的高低温一体化温度控制系统进行缓慢升温至80℃,并控制在该温度下回流、搅拌反应4h得到双(5-氯代-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(iii-3)。
步骤(3):由制备的双(5-氯代-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(iii-3)制备双(5-羟基-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(iv-3)
将水稀释在乙二醇二甲醚溶剂中,控制两者的体积比为8:100;打开步骤(1)的反应装置所连接的高低温一体化温度控制系统将反应体系的温度控制在-20℃,通过液体计量装置缓慢的将含1.04mol水的乙二醇二甲醚溶液滴加到步骤(1)的反应装置中,并向反应体系中加入1.04mol的吡啶作为酸吸收剂,控制反应体系在-20℃的低温下搅拌反应0.5h,得到双(5-羟基-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(iv-3)。经过过滤、蒸馏、萃取后得到双(5-羟基-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(iv-3)。
步骤(4):由制备的双(5-羟基-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(iv-3)制备双(5-钠氧基-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(v-3)
将由高低温一体化温度控制系统、强力搅拌系统、冷凝回流系统、真空系统、反应釜组成的反应装置经抽真空、氮气置换的方式除去整套反应装置中的空气后,在氮气保护下,将0.5mol的双(5-羟基-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(iv-3)、1l经除水干燥的乙二醇二甲醚加入到反应釜中,经搅拌溶解;使用高低温一体化温度控制系统的低温制冷功能将该双(5-羟基-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(iv-3)的乙二醇二甲醚溶液冷却到-10℃;将含乙炔钠为1.025mol的乙炔钠溶液使用加料泵在氮气保护下缓慢的滴加到反应釜中;在乙炔钠溶液滴加完毕后,关闭高低温一体化温度控制系统的低温制冷功能,使得反应装置的温度缓慢的升高到室温后继续搅拌反应0.5h得到活性中间体—双(5-钠氧基-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(v-3)。
步骤(5):由制备的双(5-钠氧基-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(v-3)制备双(5-苯氧基-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(vi-3)
通过液体计量装置缓慢的将1.04mol的碘苯滴加到步骤(4)的反应装置中,滴加完毕后,打开反应装置所连接的高低温一体化温度控制系统进行缓慢升温至80℃,并控制在该温度下回流、搅拌反应4h得到双(5-苯氧基-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(vi-3)。经过过滤、蒸馏、萃取后得到双(5-苯氧基-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(vi-3)。
步骤(6):由制备的双(5-苯氧基-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(vi-3)制备双(5-(硝基苯)-氧基-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(vii-3)
将0.5mol双(5-苯氧基-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(vi-3)加入由高低温一体化温度控制系统、强力搅拌系统、冷凝回流系统、真空系统、反应釜组成的反应装置中,开动搅拌,加入1l正己烷溶解;开动高低温一体化温度控制系统的制冷,将反应装置的温度冷却到-10℃后,缓慢的滴加浓硫酸和浓硝酸的混合溶液(浓硫酸和浓硝酸的体积比为1:2),控制所加入到混合酸液中的硝酸的物质的量为1.08mol,滴加完成后关闭高低温一体化温度控制系统的制冷,使得反应体系的温度缓慢的升到室温,在室温下搅拌反应6h后,得到含双(5-(硝基苯)-氧基-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(vii-3)的产物。将反应产物缓慢的加入1l冰水混合物中,再使用乙二醇二甲醚萃取,然后用饱和碳酸氢钾溶液洗涤有机相至中性,将有机相使用无水硫酸镁干燥后蒸馏,得到双(5-(硝基苯)-氧基-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(vii-3)。
步骤(7):由制备的双(5-(硝基苯)-氧基-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(vii-3)制备双(5-(氨基苯)-氧基-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(a-3)
将0.5mol双(5-(硝基苯)-氧基-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(vii-3)加入由加热控制系统、强力搅拌系统、冷凝回流系统、真空系统、反应釜组成的反应装置中,开动搅拌,加入1l二乙二醇二甲醚溶解;加入3mol水合肼还原体系后开动加热控制系统,将反应装置的温度升高到110℃,搅拌反应6h后,得到双(5-(氨基苯)-氧基-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(a-3)的产物。向反应产物中加入水1l后使用正己烷萃取,向水相中加入碳酸钠调节ph>10,并用正己烷萃取后合并有机相,然后使用无水硫酸镁干燥后蒸馏,得到双(5-(氨基苯)-氧基-1,3,5-三甲基-1,3,5-三苯基三硅氧基)对位碳硼烷(a-3)。
化合物(a-3)的1h-nmr谱图、13c-nmr谱图、11b-nmr谱图、29si-nmr谱图见图10-13。
实施例4双((氨基苯)-氧基二甲基硅基)邻位碳硼烷(a-1)作为环氧树脂的交联剂
将实施例1制备得到的双((氨基苯)-氧基二甲基硅基)邻位碳硼烷(a-1)作为商品化环氧树脂f-51的交联剂,与常用的4,4’-二氨基二苯基甲烷相比较,两者可在相同的工艺条件下进行交联(阶段式升温120℃、4h;140℃、2h;180℃、2h),使用量为环氧树脂与交联剂的质量比为100:40。在空气气氛下,热失重分析表明,使用双((氨基苯)-氧基二甲基硅基)邻位碳硼烷(a-1)作为交联剂的环氧树脂体系的起始分解温度为350℃,在800℃时的残重为80%,而使用4,4’-二氨基二苯基甲烷作为交联剂的环氧树脂体系的起始分解温度为300℃,在800℃时的残重不足30%。
可见与现有技术相比,由本发明化合物(a-1)作为交联剂制备的聚合物具有更好的耐高温性能。
实施例5双(3-(氨基苄)-氧-1,1,3,3-四苯基二硅氧基)间位碳硼烷(a-2)作为聚酰亚胺树脂单体
将实施例2制备得到的双(3-(氨基苄)-氧-1,1,3,3-四苯基二硅氧基)间位碳硼烷(a-2)作为聚酰亚胺树脂合成制备的二胺单体,与二氨基二苯醚二胺单体做比较,两者在相同的工艺条件下(140℃、4h)与均苯四酸二酐反应,均使用降冰片烯二酸酐作为封端剂,制备出聚酰亚胺树脂。在空气气氛下,使用双(3-(氨基苄)-氧-1,1,3,3-四苯基二硅氧基)间位碳硼烷(a-2)作为二胺单体制备的聚酰亚胺树脂的起始分解温度为540℃,在800℃时的残重为70%,使用二氨基二苯醚作为二胺制备的聚酰亚胺树脂的起始分解温度为460℃,在800℃时的残重不足20%。
可见与现有技术相比,由本发明化合物(a-2)作为单体制备的聚合物具有更好的耐高温性能。
以上,对本发明的实施方式进行了说明。但是,本发明不限定于上述实施方式。凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!