点击化学和去点击化学在药物定量合成和释放中的应用的制作方法
本发明涉及点击化学与去点击化学的应用领域,具体涉及一种点击化学和去点击化学在药物定量合成和释放中的应用。
背景技术
点击化学(clickchemistry),又译为“链接化学”、“动态组合化学”和“速配接合组合式化学”,是由美国化学家kbsharpless于2001年率先引入的一个有机合成的概念,其反应过程正如点击鼠标一样简单、高效、实用、可控,主旨是在温和的条件下,通过不同单元的拼接高选择性的形成碳-杂原子键,快速、精确的合成各种目标分子。在绿色合成和原子经济时代的背景下,clickchemistry的概念在化学合成领域做出了很大的贡献,其在药物开发和生物医用材料等诸多领域已得到广泛应用。去点击化学(declickchemistry)是由美国化学家ericv.anslyn在2016年类比sharpless定义clickchemistry的因素所提出的概念,主旨是使用温和的商业化试剂定量的还原点击反应的最初的耦合单元。到目前为止,虽然clickchemistry已得到广泛的应用,但是同时利用clickchemistry和declickchemistry实现药物的快速定量组装和定量释放还没有被报道。
技术实现要素:
本发明提出了一种点击化学和去点击化学在药物定量合成和释放中的应用,基于模板分子4-氯-7-硝基苯并-2-氧杂-1,3-二唑(nbd-cl)利用clickchemistry和declickchemistry实现了n-乙酰-l-半胱氨酸(nac)、谷胱甘(gsh)、甲硫咪唑、硫普罗宁、卡托普利、巯嘌呤等含有巯基药物分子的定量组装和释放,并利用该过程中的荧光变化监测了含巯基药物的组装和释放过程。
实现本发明的技术方案是:点击化学和去点击化学在药物定量合成和释放中的应用,以7-硝基苯呋咱类化合物或bodipy衍生物为模板分子,应用点击化学和去点击化学,实现含巯基药物分子的定量组装和定量释放。
所述7-硝基苯呋咱类化合物的结构式如下:
所述bodipy衍生物的结构式如下:
其中r为
所述含巯基药物分子为n-乙酰-l-半胱氨酸、谷胱甘肽、甲硫咪唑、硫普罗宁、卡托普利、巯嘌呤等。
应用点击化学,将nbd-cl与含巯基药物分子在pb缓冲溶液中反应,pb缓冲溶液的浓度为10mm,ph为7.4,nbd-cl与含巯基药物分子的当量比为1:1,得到定量组装产物。
应用去点击化学,将定量组装产物在亲核试剂存在下重新定量释放,得到含巯基药物分子。
所述亲核试剂为半胱氨酸、同型半胱氨酸、二硫苏糖醇或2,2’-(1,2-乙二基双氧代)双乙硫醇。
所述pb缓冲溶液中含有浓度为1-5mm的阳离子表面活性剂或脂质体。
所述阳离子表面活性剂为十六烷基三甲基溴化铵、十二烷基三甲基溴化铵、二棕榈酰磷脂酰胆碱(dppc)、二棕榈酰磷脂酰乙醇胺(dppe)或二硬脂酰磷脂酰胆碱(dspc)。
利用荧光变化监测药物的定量组装和定量释放。
以nbd-cl为模板分子,实现了药物分子n-乙酰-l-半胱氨酸(nac)、谷胱甘肽(gsh)、甲硫咪唑、硫普罗宁、卡托普利、巯嘌呤等含有巯基的药物的定量组装,然后利用半胱氨酸(cys)和同型半胱氨酸(hcy)实现了药物分子nac和gsh的定量释放,并应用该过程中的荧光变化监测了nac和gsh的组装和释放过程。其过程如下图所示:
上述应用具体包括:
测试nbd-cl储存液在未加入ctab的情况下加入与ndb-cl同等当量的gsh其吸收光谱随时间的变化。
吸收光谱的变化为:343nm处的吸收强度逐渐下降,421nm处的吸收强度逐渐上升,30min后反应未达到平衡。
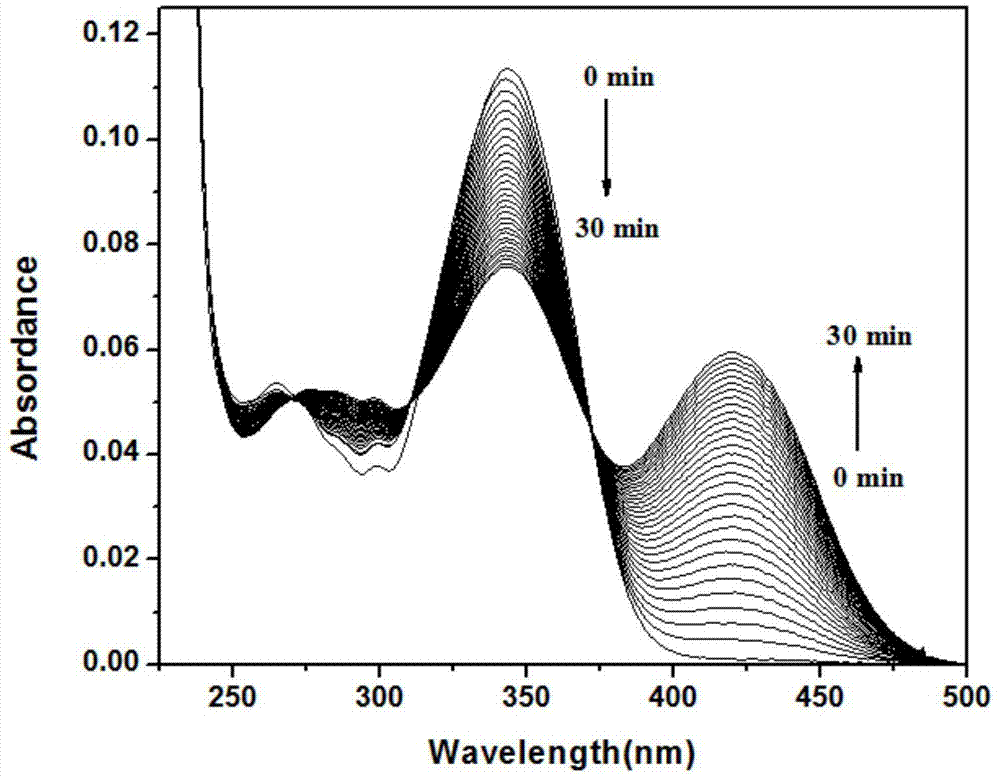
测试nbd-cl储存液在加入ctab的情况下加入与ndb-cl同等当量的gsh其吸收光谱随时间的变化。
吸收光谱的变化为:343nm处的吸收强度逐渐下降,421nm处的吸收强度逐渐上升,3min后反应达到平衡。
测试nbd-cl储存液在加入ctab的情况下加入与ndb-cl同等当量的cys其吸收光谱随时间的变化。
吸收光谱的变化为:343nm处的吸收强度逐渐下降,470nm处的吸收强度逐渐上升,3min后反应达到平衡。
测试nbd-cl储存液在加入ctab的情况下加入与ndb-cl同等当量的hcy其吸收光谱随时间的变化。
吸收光谱的变化为:343nm处的吸收强度逐渐下降,470nm处的吸收强度逐渐上升,10min后反应达到平衡。
测试nbd-cl储存液在加入ctab的情况下加入与ndb-cl同等当量的nac其吸收光谱随时间的变化。
吸收光谱的变化为:343nm处的吸收强度逐渐下降,421nm处的吸收强度逐渐上升,10min后反应达到平衡。
测试nbd-cl储存液在加入ctab的情况下加入与ndb-cl同等当量的gsh待反应达到平衡之后加入cys之后的吸收光谱变化。
吸收光谱的变化为:421nm处的吸收强度逐渐下降,470nm处的吸收强度逐渐上升,3min后反应达到平衡。
测试nbd-cl储存液在加入ctab的情况下加入与ndb-cl同等当量的gsh待反应达到平衡之后加入hcy之后的吸收光谱变化。
吸收光谱的变化为:421nm处的吸收强度逐渐下降,470nm处的吸收强度逐渐上升,3min后反应达到平衡。
测试nbd-cl储存液在加入ctab的情况下加入与ndb-cl同等当量的nac待反应达到平衡之后加入cys之后的吸收光谱变化。
吸收光谱的变化为:421nm处的吸收强度逐渐下降,470nm处的吸收强度逐渐上升,10min后反应达到平衡。
测试nbd-cl储存液在加入ctab的情况下加入与ndb-cl同等当量的nac待反应达到平衡之后加入hcy之后的吸收光谱变化。
吸收光谱的变化为:421nm处的吸收强度逐渐下降,470nm处的吸收强度逐渐上升,10min后反应达到平衡。
测试nbd-cl储存液在加入ctab的情况下加入与ndb-cl同等当量的gsh其荧光光谱随时间的变化,荧光的激发波长为470nm。
荧光光谱的变化为:以470nm为激发波长,加入gsh之后其荧光并未发生明显变化。
测试nbd-cl储存液在加入ctab的情况下加入与ndb-cl同等当量的cys其荧光光谱随时间的变化,荧光的激发波长为470nm。
荧光光谱的变化为:以470nm为激发波长,加入cys之后543nm处的荧光逐渐增强。
测试nbd-cl储存液在加入ctab的情测试nbd-cl储存液在加入ctab的情况下加入与ndb-cl同等当量的gsh待反应达到平衡之后加入cys之后的荧光光谱变化,荧光的激发波长为470nm。
荧光光谱的变化为:以470nm为激发波长,加入cys之后543nm处的荧光逐渐增强。
测试nbd-cl储存液在加入ctab的情况下加入与ndb-cl同等当量的nac其荧光光谱随时间的变化,荧光的激发波长为470nm。
荧光光谱的变化为:以470nm为激发波长,加入nac之后其荧光并未发生明显变化。
测试nbd-cl储存液在加入ctab的情况下加入与ndb-cl同等当量的nac待反应达到平衡之后加入cys之后的荧光光谱变化,荧光的激发波长为470nm。
荧光光谱的变化为:以470nm为激发波长,加入cys之后543nm处的荧光逐渐增强。
本发明的有益效果是:(1)模板分子可以与含巯基药物分子在表面活性剂的存在下快速1:1的反应。(2)模板分子与含巯基药物分子组装之后的分子可以在亲核试剂的存在下快速的发生declick-chemistry释放含巯基药物分子。(3)在含巯基药物分子释放过程中存在荧光上的变化,可以方便的利用其荧光变化监测药物分子的释放。(4)以上反应均在磷酸缓冲溶液中进行,具有反应条件温和,原料和反应试剂易得,反应速度快,反应产率高等特点,符合原子经济。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1是在pb缓冲溶液(10mm,ph=7.4)体系中,10μmnbd-cl与10μmgsh反应时,吸收光谱随时间的变化图。
图2是在pb缓冲溶液(10mm,ph=7.4,1mmctab)体系中,10μmnbd-cl与10μmgsh反应时,吸收光谱随时间的变化图。
图3是在pb缓冲溶液(10mm,ph=7.4,1mmctab)体系中,10μmnbd-cl与10μmcys反应时,吸收光谱随时间的变化图。
图4是在pb缓冲溶液(10mm,ph=7.4,1mmctab)体系中,10μmnbd-cl与10μmhcy反应时,吸收光谱随时间的变化图。
图5是在pb缓冲溶液(10mm,ph=7.4,1mmctab)体系中,10μmnbd-cl与10μmnac反应时,吸收光谱随时间的变化图。
图6是在pb缓冲溶液(10mm,ph=7.4,1mmctab)体系中,10μmnbd-cl与10μmgsh反应平衡之后加入100μmcys时,吸收光谱随时间的变化图。
图7是在pb缓冲溶液(10mm,ph=7.4,1mmctab)体系中,10μmnbd-cl与10μmgsh反应平衡之后加入100μmhcy时,吸收光谱随时间的变化图。
图8是在pb缓冲溶液(10mm,ph=7.4,1mmctab)体系中,10μmnbd-cl与10μmnac反应平衡之后加入100μmcys时,吸收光谱随时间的变化图。
图9是在pb缓冲溶液(10mm,ph=7.4,1mmctab)体系中,10μmnbd-cl与10μmnac反应平衡之后加入100μmhcy时,吸收光谱随时间的变化图。
图10是在pb缓冲溶液(10mm,ph=7.4,1mmctab)体系中,10μmnbd-cl与10μmgsh反应时,以470nm为激发波长,荧光光谱随时间的变化图。
图11是在pb缓冲溶液(10mm,ph=7.4,1mmctab)体系中,10μmnbd-cl与10μmcys反应时,以470nm为激发波长,荧光光谱随时间的变化图。
图12是在pb缓冲溶液(10mm,ph=7.4,1mmctab)体系中,10μmnbd-cl与10μmnac反应时,以470nm为激发波长,荧光光谱随时间的变化图。
图13是在pb缓冲溶液(10mm,ph=7.4,1mmctab)体系中,10μmnbd-cl与10μmgsh反应平衡之后加入100μmcys时,以470nm为激发波长,荧光光谱随时间的变化图。
图14是在pb缓冲溶液(10mm,ph=7.4,1mmctab)体系中,10μmnbd-cl与10μmnac反应平衡之后加入100μmcys时,以470nm为激发波长,荧光光谱随时间的变化图。
图15是在pb缓冲溶液(10mm,ph=7.4,1mmctab)体系中,10μm的nbd-cl,10μmnbd-cl与10μmgsh反应10min,10μmnbd-cl与100μm的cys反应10min,和10μmnbd-cl与10μmgsh反应10min之后加入100μm的cys反应10min的吸收光谱图。
图16是在pb缓冲溶液(10mm,ph=7.4,1mmctab)体系中,10μm的nbd-cl,10μmnbd-cl与10μmgsh反应10min,10μmnbd-cl与100μm的hcy反应10min,和10μmnbd-cl与10μmgsh反应10min之后加入100μm的hcy反应10min的吸收光谱图。
图17是在pb缓冲溶液(10mm,ph=7.4,1mmctab)体系中,10μm的nbd-cl,10μmnbd-cl与10μmnac反应10min,10μmnbd-cl与100μm的cys反应10min,和10μmnbd-cl与10μmnac反应10min之后加入100μm的cys反应10min的吸收光谱图。
图18是在pb缓冲溶液(10mm,ph=7.4,1mmctab)体系中,10μm的nbd-cl,10μmnbd-cl与10μmnac反应10min,10μmnbd-cl与100μm的hcy反应10min,和10μmnbd-cl与10μmnac反应10min之后加入100μm的hcy反应10min的吸收光谱图。
具体实施方式
下面将结合本发明实施例,对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有付出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
向2ml的pb缓冲(10mm,ph=7.4,1mmctab)体系中,加入10μl浓度为2mm的nbd-cl储备液,再加入1μl浓度为20mm的gsh,测试其吸收光谱随时间的变化,如图2所示,343nm处的吸收逐渐下降,421nm处的吸收逐渐上升,3min左右达到反应平衡。待反应达到平衡后,再加入10μl浓度为20mm的cys或hcy,如图6,7所示,421nm处的吸收逐渐下降,470nm处的吸收峰逐渐逐渐上升,3min左右达到平衡,如图15,16所示,其达到平衡时的吸收光谱与nbd-cl(10μm)加入cys或hcy(10μm)时的吸收光谱基本一致。
向2ml的pb缓冲(10mm,ph=7.4,1mmctab)体系中,加入10μl浓度为2mm的nbd-cl储备液,再加入1μl浓度为20mm的gsh,测试其荧光光谱随时间的变化,如图10所示,543nm处的荧光没有明显的变化。待反应达到平衡后,再加入10μl浓度为20mm的cys,如图13所示,543nm处的吸收峰逐渐上升,其达到平衡时的荧光光谱与nbd-cl(10μm)加入cys(10μm)时的荧光光谱基本一致。
实施例2
向2ml的pb缓冲(10mm,ph=7.4,2.5mmctab)体系中,加入10μl浓度为2mm的nbd-cl储备液,再加入1μl浓度为20mm的gsh,测试其吸收光谱随时间的变化,343nm处的吸收逐渐下降,421nm处的吸收逐渐上升,3min左右达到反应平衡。待反应达到平衡后,再加入10μl浓度为20mm的cys或hcy,421nm处的吸收逐渐下降,470nm处的吸收峰逐渐逐渐上升,3min左右达到平衡,其达到平衡时的吸收光谱与nbd-cl(10μm)加入cys或hcy(10μm)时的吸收光谱基本一致。
向2ml的pb缓冲(10mm,ph=7.4,1mmctab)体系中,加入10μl浓度为2mm的nbd-cl储备液,再加入1μl浓度为20mm的gsh,测试其荧光光谱随时间的变化,543nm处的荧光没有明显的变化。待反应达到平衡后,再加入10μl浓度为20mm的cys,543nm处的吸收峰逐渐上升,其达到平衡时的荧光光谱与nbd-cl(10μm)加入cys(10μm)时的荧光光谱基本一致。
实施例3
向2ml的pb缓冲(10mm,ph=7.4,5mmctab)体系中,加入10μl浓度为2mm的nbd-cl储备液,再加入1μl浓度为20mm的gsh,测试其吸收光谱随时间的变化,343nm处的吸收逐渐下降,421nm处的吸收逐渐上升,3min左右达到反应平衡。待反应达到平衡后,再加入10μl浓度为20mm的cys或hcy,421nm处的吸收逐渐下降,470nm处的吸收峰逐渐逐渐上升,3min左右达到平衡,其达到平衡时的吸收光谱与nbd-cl(10μm)加入cys或hcy(10μm)时的吸收光谱基本一致。
向2ml的pb缓冲(10mm,ph=7.4,1mmctab)体系中,加入10μl浓度为2mm的nbd-cl储备液,再加入1μl浓度为20mm的gsh,测试其荧光光谱随时间的变化,543nm处的荧光没有明显的变化。待反应达到平衡后,再加入10μl浓度为20mm的cys,543nm处的吸收峰逐渐上升,其达到平衡时的荧光光谱与nbd-cl(10μm)加入cys(10μm)时的荧光光谱基本一致。
以下实施例其余条件同实施例1。
nbd-cl加入同等当量的gsh然后加入cys或hcy的光谱研究
(1)吸收光谱研究
向2ml的pb缓冲(10mm,ph=7.4,1mmctab)体系中,加入10μl浓度为2mm的nbd-cl储备液,再加入1μl浓度为20mm的gsh,测试其吸收光谱随时间的变化,如图2所示,343nm处的吸收逐渐下降,421nm处的吸收逐渐上升,3min左右达到反应平衡。待反应达到平衡后,再加入10μl浓度为20mm的cys或hcy,如图6,7所示,421nm处的吸收逐渐下降,470nm处的吸收峰逐渐逐渐上升,3min左右达到平衡,如图15,16所示,其达到平衡时的吸收光谱与nbd-cl(10μm)加入cys或hcy(10μm)时的吸收光谱基本一致,其结果表明,10μm的nbd-cl与10μm的gsh,按照当量比1:1反应完全,无其它副产物且反应迅速,向反应完全的溶液中加入100μm的cys或hcy,待反应平衡后又重新释放出10μm的gsh。
(2)荧光光谱研究
向2ml的pb缓冲(10mm,ph=7.4,1mmctab)体系中,加入10μl浓度为2mm的nbd-cl储备液,再加入1μl浓度为20mm的gsh,测试其荧光光谱随时间的变化,如图10所示,543nm处的荧光没有明显的变化。待反应达到平衡后,再加入10μl浓度为20mm的cys,如图13所示,543nm处的吸收峰逐渐上升,其达到平衡时的荧光光谱与nbd-cl(10μm)加入cys(10μm)时的荧光光谱基本一致,其结果表明,可以利用荧光的变化来定量监测gsh的释放量。
2.nbd-cl加入同等当量的nac然后加入cys或hcy的光谱研究
(1)吸收光谱研究
向2ml的pb缓冲(10mm,ph=7.4,1mmctab)体系中,加入10μl浓度为2mmnbd-cl储备液,再加入1μl浓度为20mm的nac,测试其吸收光谱随时间的变化,如图5所示,343nm处的吸收逐渐下降,421nm处的吸收逐渐上升,3min左右达到反应平衡。待反应达到平衡后,再加入10μl浓度为20mm的cys或hcy,如图8,9所示,421nm处的吸收逐渐下降,470nm处的吸收峰逐渐逐渐上升,3min左右达到平衡,如图17,18所示,其达到平衡时的吸收光谱与nbd-cl(10μm)加入cys或hcy(10μm)时的吸收光谱基本一致,其结果表明,其结果表明,10μm的nbd-cl与10μm的nac,按照当量比1:1反应完全,无其它副产物且反应迅速,向反应完全的溶液中加入100μm的cys或hcy,待反应平衡后又重新释放出10μm的nac。
(2)荧光光谱研究
向2ml的pb缓冲(10mm,ph=7.4,1mmctab)体系中,加入10μl浓度为2mm的nbd-cl储备液,再加入1μl浓度为20mm的nac,测试其荧光光谱随时间的变化,如图12所示,543nm处的荧光没有明显的变化。待反应达到平衡后,再加入10μl浓度为20mm的cys,如图14所示,543nm处的吸收峰逐渐上升,其达到平衡时的荧光光谱与nbd-cl(10μm)加入cys(10μm)时的荧光光谱基本一致,其结果表明,可以利用荧光的变化来定量监测nac的释放量。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!