一种有机聚合物、其合成方法及在制备超高性能锂电池负极上的应用与流程
本发明涉及本发明是一种高性能的锂离子电池负极材料,尤其是涉及一种有机聚合物、其合成方法及在制备超高性能锂电池负极上的应用。
背景技术
传统化石能源的枯竭及环境污染使得人们对清洁能源的需求越来越大。近几年来,作为绿色可持续能源的杰出代表,锂离子电池受到的关注日益增加。从宏观角度来说,锂电池的研究寄托着国家能源振兴战略的重任。然而就目前的发展水平来说,锂离子的性能指标与实际应用要求还有很大差距。比如,广泛应用的锂离子负极材料—石墨材料理论容量上限仅仅为372mah/g,而且也大电流充放电性能也不高。因此制备高容量、大电流充放电的锂电池负极材料是近几年研究工作的核心问题之一,是解决目前锂离子电池瓶颈问题的关键。
为解决上述问题,人们进行一系列的研究,关注点多集中在组合材料领域。申请号为200980147980.5的中国专利提供了充放电循环特性优异的锂二次电池负极用炭材料、锂二次电池负极及使用该负极的锂二次电池。本发明的锂二次电池负极用炭材料的特征在于,包含:复合颗粒,其由能吸贮·释放锂离子的含硅颗粒和包围该含硅颗粒的树脂炭材料构成,所述含硅颗粒含有硅的合金、氧化物、氮化物或碳化物;和网状结构体,其由结合于该复合颗粒的表面且包围该复合颗粒的纳米纤维和/或纳米管构成,其中,该网状结构体含有硅。申请号为201710540524.3的中国专利开了一种复合型锂电池负极材料及其制备方法和锂电池负极、锂电池,所述复合型锂电池负极材料的制备方法,包括以下步骤:将含有锡源、硫源、石墨烯衍生物、表面活性剂和碱源的溶液在160~240℃下进行水热反应,得到复合型锂电池负极材料。这两篇专利从能源利用和循环方面为锂电池的发展提供新的思路,但所得电极材料所得电池充放电的速度和容量没有明显提升。
技术实现要素:
有鉴于此,本发明的目的是针对现有技术的不足和锂离子电池发展的需要,提供一种超高性能负极材料的制备方法。该材料在200mag-1充放电速率下循环400圈容量保持1700mahg-1以上,在1000mag-1的充放电速率下循环400圈容量不低于760mahg-1。
为达到上述目的,本发明采用以下技术方案:
一种聚合物pat,是由2,6二氨基蒽醌与三聚氯氰在碱得作用下反应,其结构如下,
所述的聚合物的制备方法,包括如下步骤:将1当量2,6二氨基蒽醌、0.5-3.0当量的三聚氯氰和1.0-20当量的碱加入到溶剂中,加热,n2保护下,反应24-100h,经纯化处理,得聚合物;为提高聚合物的聚合度,反应溶剂采用高沸点溶剂,碱采用碳酸盐;其反应式如下:
进一步地,所述碱为碳酸钠、碳酸钾。
进一步地,所述溶剂为dmf或者dmso,反应温度为80-140摄氏度。
进一步地,所述纯化方法为索氏提取,所用溶剂为乙醇、乙腈与二氯甲烷的混合溶剂。
所述聚合物在制备超高性能锂电池负极上的应用,包括如下步骤:pat、导电炭黑、粘合剂按照一定的重量比混合,制备成锂电池负极材料;具体为pat:炭黑:粘结剂60:25:15-80:10:10;所述粘结剂为pvdf。
本发明的有益效果是:
本发明提供了一种新的聚合物pat,并对该聚合物的结构、性能进行详细的表征和研究,为该聚合物的应用奠定了基础。
本发明提供了一种应用聚合物pat制备大电流充放电下,高比容量锂离子电池负极材料,该材料易于合成,成本低廉,性能稳定。作为负极材料,在200mag-1充放电速率下循环400圈容量保持1700mahg-1以上;在1a/g大电流充放电下,容量高于760mahg-1。有望代替传统的石墨负极材料,制备高性能锂离子电池。
附图说明
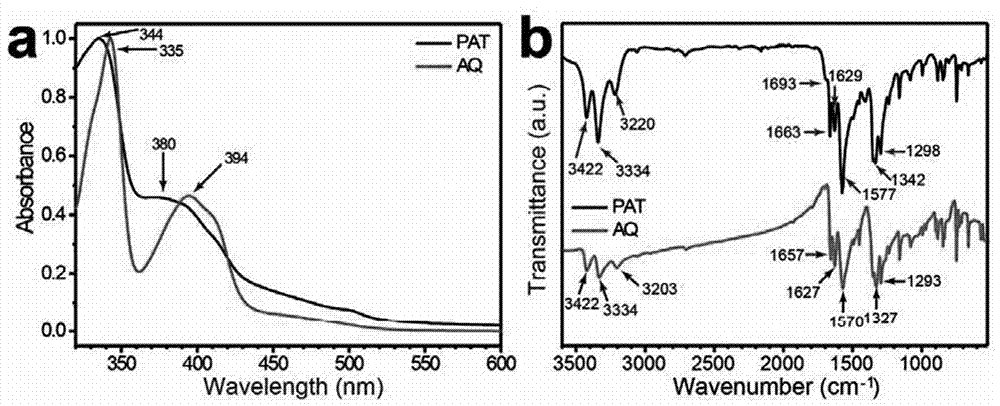
图1为聚合物的紫外吸收、红外光谱图:(a)紫外吸收光谱(dmf溶液中)和(b)aq(red)andpat(black)的ft-ir光谱;
图2为聚合物的(n2)吸附脱附等温线:(a)n2吸附脱附曲线和(b)聚合物pat的孔径分布;
图3为聚合物的孔径分布:pat的充放电性能(a)200mag-1下的gdc曲线,(b)200mag-1的充放电性能,(c)1ag-1的充放电性能和(d)倍率性能。
具体实施方式
下面结合附图和实施例对本发明作进一步描述。
聚合物的制备方法
实施例1
氮气保护下,2,6二氨基蒽醌aq(714mg,3.0mmol)三聚氯氰tct(274.5mg,1.5mmol)和na2co3(0.318g,3.0mmol)加入到三口瓶中,氮气置换3次后,加入15mln,n-二甲基甲酰胺(dmf).该混合物在80℃反应10h后,升温至140℃并保温反应14-24h.待反应完成后,将至室温,将反应液倒入100ml二次水中并搅拌30分钟.沉淀滤出后,用二次水和乙醇洗涤,然后使用乙醇/乙腈、二氯甲烷作为提洗液进行索氏提取2天。经55℃真空干燥12h后,得到黄色产品603.5mg,产率,70.1%.1hnmr:(400mhz,dmso-d6)δ10.83-10.77and6.67-6.53forn-h,(m,2h),8.44-6.83foraromatic-h(m,6h);13cnmr:(100mhz,dmso-d6)δ182.26,181.09,160.46,154.84,154.60,143.61,143.43,135.88,135.13,129.68,129.30,128.62,128.48,128.29,123.76,122.95,121.46,117.85,116.98,116.07,115.95,109.78,109.68.
实施例2
反应方程式如实施例1所示:
氮气保护下,2,6二氨基蒽醌aq(714mg,3.0mmol)三聚氯氰tct(366mg,2.0mmol)和na2co3(1.06g,10.0mmol)加入到三口瓶中,氮气置换3次后,加入15mln,n-二甲基甲酰胺(dmf).该混合物在100℃反应50h后,升温至140℃并保温反应50h.待反应完成后,将至室温,将反应液倒入150ml二次水中并搅拌30分钟.沉淀滤出后,用二次水和乙醇洗涤,然后使用乙醇/乙腈、二氯甲烷作为提洗液进行索氏提取2天。经55℃真空干燥12h后,得到黄色产品560.0mg,产率:65.0%.1hnmr:(400mhz,dmso-d6)δ10.83-10.77and6.67-6.53forn-h,(m,2h),8.44-6.83foraromatic-h(m,6h);13cnmr:(100mhz,dmso-d6)δ182.26,181.09,160.46,154.84,154.60,143.61,143.43,135.88,135.13,129.68,129.30,128.62,128.48,128.29,123.76,122.95,121.46,117.85,116.98,116.07,115.95,109.78,109.68.
实施例3
反应方程式如实施例1所示:
氮气保护下,2,6二氨基蒽醌aq(714mg,3.0mmol)三聚氯氰tct(1.647g,9.0mmol)和k2co3(6.36g,60.0mmol)加入到三口瓶中,氮气置换3次后,加入15mln,n-二甲基甲酰胺(dmf).该混合物在100℃反应20h后,升温至140℃并保温反应24h.待反应完成后,将至室温,将反应液倒入100ml二次水中并搅拌30分钟.沉淀滤出后,用二次水和乙醇洗涤,然后使用乙醇/乙腈、二氯甲烷作为提洗液进行索氏提取2天。经55℃真空干燥12h后,得到黄色产品600.0mg,产率70.0%.1hnmr:(400mhz,dmso-d6)δ10.83-10.77and6.67-6.53forn-h,(m,2h),8.44-6.83foraromatic-h(m,6h);13cnmr:(100mhz,dmso-d6)δ182.26,181.09,160.46,154.84,154.60,143.61,143.43,135.88,135.13,129.68,129.30,128.62,128.48,128.29,123.76,122.95,121.46,117.85,116.98,116.07,115.95,109.78,109.68.
实施例4
反应方程式如实施例1所示:
氮气保护下,2,6二氨基蒽醌aq(714mg,3.0mmol)三聚氯氰tct(366mg,2.0mmol)和na2co3(0.318g,3.0mmol)加入到三口瓶中,氮气置换3次后,加入15ml二甲基亚砜(dmso).该混合物在100℃反应20h后,升温至140℃并保温反应24h.待反应完成后,将至室温,将反应液倒入100ml二次水中并搅拌30分钟.沉淀滤出后,用二次水和乙醇洗涤,然后使用乙醇/乙腈、二氯甲烷作为提洗液进行索氏提取2天。经55℃真空干燥12h后,得到黄色产品526mg,产率61.1%.1hnmr:(400mhz,dmso-d6)δ10.83-10.77and6.67-6.53forn-h,(m,2h),8.44-6.83foraromatic-h(m,6h);13cnmr:(100mhz,dmso-d6)δ182.26,181.09,160.46,154.84,154.60,143.61,143.43,135.88,135.13,129.68,129.30,128.62,128.48,128.29,123.76,122.95,121.46,117.85,116.98,116.07,115.95,109.78,109.68.
实施例5
电极复合物的制备
电极复合物的制备:称取60wt%的pat,25wt%的导电炭黑,15wt%的pvdf,加入几滴n-甲基吡咯烷酮(nmp)后,研磨30分钟,然后该均匀的浆料涂在20μm厚的铜箔上,真空60℃干燥24h。然后以锂片为对电极,组装成cr2032电池。电解液采用1.0mlipf6(碳酸乙烯酯:碳酸二甲酯=1:1),充放电测试范围为0.01-3.0v。
所述化合物的状态和性质表征:
紫外-可见吸收采用u-4100光谱仪(uv-vis-nirhitachispectrometer),红外光谱使用thermoscientificnicoletis5傅立叶红外光谱仪,扫描范围为700-3500cm-1,见附图1;可以看出,聚合后,紫外可见光谱蓝移,这可能与聚合物骨架上aq单元降低了的ict过程有关。红外光谱3422cm-1and3342cm-1处的峰,对应于pat结构上的不对称及对称性的n-h键振动,相比aq分子,pat的3422cm-1处吸收更强,可能是三嗪的引入d带来了更大的不对称有关。pat的1693cm-1处肩缝也明显增强,并且c=o伸缩振动峰朝着更高能量方向移动(从aq小分子的1627cm-1、1657cm-1处到pat的1629cm-1和1663cm-1处).aq的1570cm-1和pat的1577cm-1处吸收均属于蒽醌骨架的吸收。
附图2,3氮气吸附脱附曲线使用micromeriticsasap2020仪器于77k测试,,样品脱气条件:120℃,6小时;通过附图2可以看出,该吸附脱附曲线为第四种曲线,说明该聚合物是典型的介孔材料,从2b中可以看出,材料的微孔、介孔占主导,brunauer–emmett–teller方法测得的比表面积高达140m2/g,总孔容积为(vtol)0.62cm3g-1,显然这种高的比表面积和孔状结构有利于锂离子的传输,并提供了大量的电化学反应位点。
从附图3a可以看出,该聚合物的电位比锂电位要高一些,因此作为负极能够明显的避免出现锂金属的支晶,从而带来电池的稳定性。附图3b、3c及3d可以看出,该聚合物具有很大的比容量,并且倍率性能优良。
最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,本领域普通技术人员对本发明的技术方案所做的其他修改或者等同替换,只要不脱离本发明技术方案的精神和范围,均应涵盖在本发明的权利要求范围当中。
- 还没有人留言评论。精彩留言会获得点赞!