重组猪伪狂犬病毒rPRVHN2012-TK-/gE-/gI-及其构建方法和应用与流程
本发明涉及病毒构建技术领域,具体涉及重组猪伪狂犬病毒rprvhn2012-tk-/ge-/gi-及其构建方法和应用。
背景技术
伪狂犬病(pseudorabies,pr),即奥耶兹氏病(aujeszky’sdisease),其病原是伪狂犬病病毒(pseudorabiesvirus,prv),牛、羊、猪等多种家畜及野生动物均可患该病。pr的主要症状为发热、奇痒、脑脊髓炎、神经及繁殖系统障碍,是一种急性传染病,而且目前尚无研发出可成功治疗pr的药物,只能在临床上采用接种如prvbartha-k61等弱毒疫苗来对pr进行防控。但2011年后,我国多个省份中已免疫过伪狂犬疫苗的猪群中出现了新一轮pr疫情的大流行。随后许多研究也陆续证实了市场上常接种的bartha-k61弱毒疫苗株对当前流行的prv变异株已不能提供完全有效的保护力,这一轮以prv流行变异株引发的pr疫情使我国的养猪业面临巨大的风险,因此一种可针对当前流行变异株的有效疫苗急待开发。
技术实现要素:
本发明的目的在于提供重组猪伪狂犬病毒rprvhn2012-tk-/ge-/gi-及其构建方法和应用。本发明提供的病毒安全性和免疫效力均很高,能有效应用于当前prv流行防控工作的疫苗制备过程中。
本发明提供了重组猪伪狂犬病毒rprvhn2012-tk-/ge-/gi-,保藏编号为cctccno:v201748。
本发明还提供了上述技术方案所述病毒的构建方法,包括以下步骤:
1)分别扩增疱疹病毒科猪疱疹病毒i型猪伪狂犬病毒prv/hn2012的tk基因两侧序列克隆至puc-19载体,得到第一转移质粒puc-tklr,再将egfp基因表达盒克隆至转移质粒puc-tklr上,获得第二转移质粒puc-tklre;所述egfp基因表达盒克隆的位置在tk基因两侧序列的中间;
2)将重组猪伪狂犬病毒rprvhn2012-ge-/gi-接种于细胞中,吸附1~2h后,得到经重组猪伪狂犬病毒rprvhn2012-ge-/gi-感染的细胞,将步骤1)得到的第二转移质粒puc-tklre转染于经重组猪伪狂犬病毒rprvhn2012-ge-/gi-感染的细胞中,通过空斑纯化获得表达荧光蛋白的过渡病毒rprvhn2012-ge-/gi-/tk--egfp+;
3)将步骤2)得到的过渡病毒rprvhn2012-ge-/gi-/tk--egfp+接种于细胞中,吸附1~2h后,得到经过渡病毒rprvhn2012-ge-/gi-/tk--egfp+感染的细胞,将所述步骤1)得到的第一转移质粒puc-tklr转染于经过渡病毒rprvhn2012-ge-/gi-/tk--egfp+感染的细胞中,通过空斑纯化去除egfp基因表达盒,得到重组猪伪狂犬病毒rprvhn2012-tk-/ge-/gi-;
所述疱疹病毒科猪疱疹病毒i型猪伪狂犬病毒prv/hn2012的保藏编号为cctccno:v201314;
所述重组猪伪狂犬病毒rprvhn2012-ge-/gi-的保藏编号为cctccno:v201749。
优选的是,步骤1)所述tk基因两侧序列的核苷酸长度独立地为750~1500bp。
优选的是,步骤1)所述egfp基因表达盒的核苷酸序列由以下基因片段依次排布组成:cmv、mcs、egfp和sv40polya尾。
优选的是,步骤2)和步骤3)接种用细胞包括st细胞。
优选的是,所述st细胞待培养至细胞融合70%时,用于接种。
优选的是,步骤2)所述空斑纯化的次数为4~7次,在蓝光条件下观察到所有病毒蚀斑呈绿色荧光时表明纯化完全。
优选的是,步骤3)所述空斑纯化的次数为4~7次,在蓝光条件下观察到所有病毒蚀斑不呈现绿色荧光时表明纯化完全。
本发明还提供了上述技术方案所述重组猪伪狂犬病毒rprvhn2012-tk-/ge-/gi-或上述技术方案所述制备方法制备得到的重组猪伪狂犬病毒rprvhn2012-tk-/ge-/gi-在制备猪伪狂犬病病毒疫苗中的应用。
本发明提供了重组猪伪狂犬病毒rprvhn2012-tk-/ge-/gi-。本发明提供的重组病毒安全性高,免疫效力好。试验结果表明,rprvhn2012-tk-/ge-/gi-免疫组小鼠在试验过程中没有出现任何不良反应,病毒致弱情况理想;间接elisa结果表明rprvhn2012-tk-/ge-/gi-免疫组在免疫过程中产生了较高滴度的prv抗体,中和抗体水平也证实了该缺失病毒保持了良好的免疫原性,此外小鼠外周血t淋巴细胞亚群的测定结果也表明该缺失病毒有效地刺激小鼠机体产生细胞免疫;小鼠的脑部病理切片也说明了弱毒株在一定程度上也阻碍了强毒株对神经组织的侵袭,发挥了一定的保护作用。总之,本发明rprvhn2012-tk-/ge-/gi-三基因缺失病毒对非靶动物小鼠是安全的,免疫后能有效并迅速产生prv特异性抗体,且能有效地激活细胞免疫,是一株有希望应用于当前prv流行防控工作的疫苗株。
附图说明
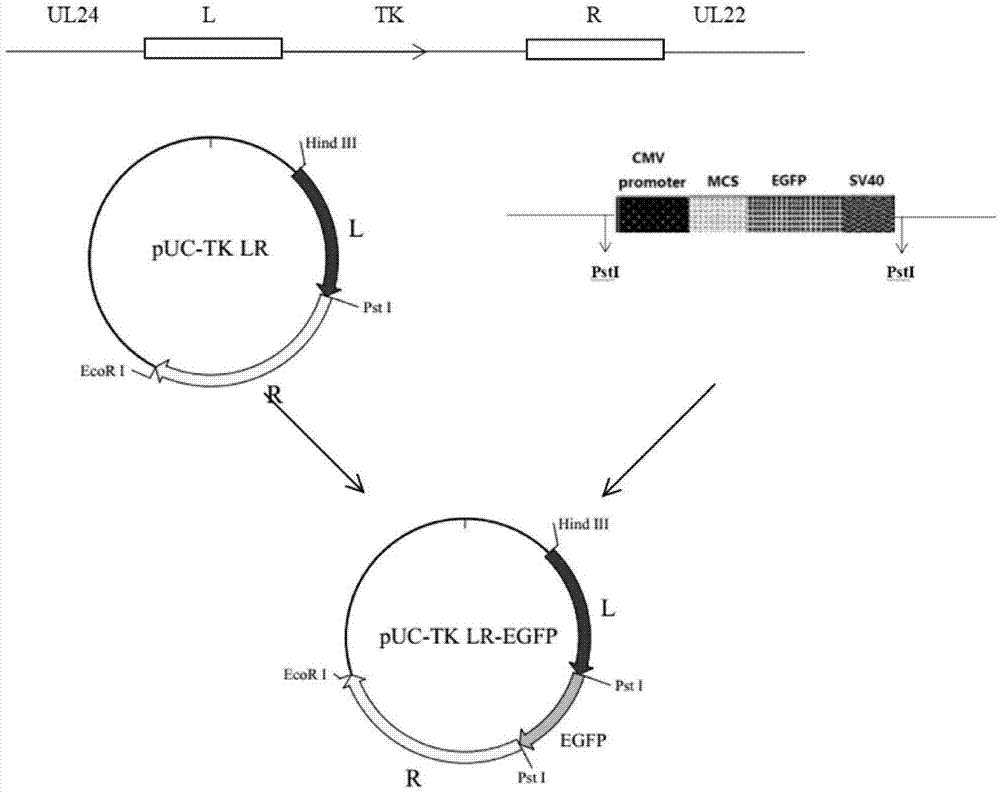
图1为本发明实施例1提供的转移质粒构建技术路线图;
图2为本发明实施例1提供的pg质粒基因结构图;
图3为本发明实施例1提供的同源臂的扩增截图;
图4为本发明实施例1提供的重组质粒pmd-tkl、pmd-tkr的酶切鉴定图;
图5为本发明实施例1提供的转移质粒的酶切鉴定图;
图6为本发明实施例1提供的pg质粒的酶切鉴定图;
图7为本发明实施例1提供的菌液pcr鉴定图;
图8为本发明实施例2提供的不同光照下观察到的蚀斑照片;
图9为本发明实施例2提供的tk缺失部分pcr鉴定结果图;
图10为本发明实施例2提供的不同光照下观察到的蚀斑照片;
图11为本发明实施例2提供的tk缺失部分pcr鉴定结果图;
图12为本发明实施例2提供的重组病毒rprvhn2012-tk-/ge-/gi-gb基因的鉴定结果图;
图13为本发明实施例2提供的缺失病毒和亲本毒株的一步生长曲线图;
图14为本发明实施例3提供的小鼠ppv抗体检测结果图;
图15为本发明实施例3提供的prv中和抗体效价水平(log2)结果图;
图16为本发明实施例3提供的小鼠外周血中cd3+、cd4+、cd8+t淋巴细胞数量的测定结果图;
图17为本发明实施例3提供的小鼠的生存曲线图;
图18为本发明实施例3提供的死亡小鼠的伤口状态结果图;
图19为本发明实施例3提供的小胶质细胞聚集图;
图20为本发明实施例3提供的小鼠的脑部病理切片结果图。
具体实施方式
本发明提供了重组猪伪狂犬病毒rprvhn2012-tk-/ge-/gi-,保藏编号为cctccno:v201748。本毒株保藏在中国典型培养物保藏中心,地址为湖北省武汉市武昌区八一路299号,武汉大学,保藏时间为2017年10月31日。tk基因的缺失可以明显降低prv的侵染力、传播力和毒力,但仍能使其保持良好的免疫原性。
本发明还提供了上述技术方案所述病毒的构建方法,包括以下步骤:
1)分别扩增疱疹病毒科猪疱疹病毒i型猪伪狂犬病毒prv/hn2012的tk基因两侧序列克隆至puc-19载体,得到第一转移质粒puc-tklr,再将egfp基因表达盒克隆至转移质粒puc-tklr上,获得第二转移质粒puc-tklre;所述egfp基因表达盒克隆的位置在tk基因两侧序列的中间;
2)将重组猪伪狂犬病毒rprvhn2012-ge-/gi-接种于细胞中,吸附1~2h后,得到经重组猪伪狂犬病毒rprvhn2012-ge-/gi-感染的细胞,将步骤1)得到的第二转移质粒puc-tklre转染于经重组猪伪狂犬病毒rprvhn2012-ge-/gi-感染的细胞中,通过空斑纯化获得表达荧光蛋白的过渡病毒rprvhn2012-ge-/gi-/tk--egfp+;
3)将步骤2)得到的过渡病毒rprvhn2012-ge-/gi-/tk--egfp+接种于细胞中,吸附1~2h后,得到经过渡病毒rprvhn2012-ge-/gi-/tk--egfp+感染的细胞,将所述步骤1)得到的第一转移质粒puc-tklr转染于经过渡病毒rprvhn2012-ge-/gi-/tk--egfp+感染的细胞中,通过空斑纯化去除egfp基因表达盒,得到重组猪伪狂犬病毒rprvhn2012-tk-/ge-/gi-;
所述疱疹病毒科猪疱疹病毒i型猪伪狂犬病毒prv/hn2012的保藏编号为cctccno:v201314,本毒株保藏在中国典型培养物保藏中心,地址为湖北省武汉市武昌区八一路299号,武汉大学,保藏时间为2013年5月28日。
所述重组猪伪狂犬病毒rprvhn2012-ge-/gi-的保藏编号为cctccno:v201749,本本毒株保藏在中国典型培养物保藏中心,地址为湖北省武汉市武昌区八一路299号,武汉大学,保藏时间为2017年10月31日。
本发明分别扩增疱疹病毒科猪疱疹病毒i型猪伪狂犬病毒prv/hn2012的tk基因两侧序列克隆至puc-19载体,得到第一转移质粒puc-tklr,再将egfp基因表达盒克隆至转移质粒puc-tklr上,获得第二转移质粒puc-tklre;所述egfp基因表达盒克隆的位置在tk基因两侧序列的中间。在本发明中,所述tk基因两侧序列的核苷酸长度独立地为750~1500bp,更优选为967~976bp,最优选分别为967bp和976bp,本发明tk基因两侧序列(左右重组同源臂)的长度范围的设置能够保证同源重组的顺利进行。本发明tk基因两侧序列分别作为左、右重组同源臂,用于后续tk基因的缺失株的构建。当所述tk基因两侧序列的核苷酸长度分别为967bp和976bp时,本发明优选设计两对特异性引物tkl/f和tkl/r、tkr/f和tkr/r(如表1所示)用于左、右重组同源臂的扩增;本发明左重组同源臂优选包括部分ul24基因和部分tk基因,大小为967bp,具体序列如下:aagcttgccttatcatccccgctccccgccgccgcccggcccggccccgcgcgcgccgcgatcgcgatcaccgccgcggcccggcgacgtactcggcgaggccgcgcacggtcgcggccatcgcgctcgcgttgccgcgcgtctgggtgcagggcaggcgcgtcacgtcgagcacgcgcatgctccgctgggccacaaacaccagcaggggcacgagcgtgatctcctcgccgcccgggggcacggcggcggcgaggaggcgcgccgagtcgcgcagctggcacagcccctcgtgccgctgcccgcgcttgctgggcgtgttgaggttccgggggaagcggcacgtcttgagctcgatgaggaagcacaggtgcgggcccgcccccagccgcaccacgcacacgcagtcggggcggcgcaccccgaggttgacttcaaaggccagggtcaaggacgccttcttaagcgtctctcggggaagcccgaagagactctcgccgtacgcggacgggtcgcgtcgcaggcgttcgtagaagcggttgtggcagcggatccccgcccggaagcgcgccgggatgcgcatcctccggatctacctcgacggcgcctacggcaccggcaagagcaccacggcccgggtgatggcgctcggcggggcgctgtacgtgcccgagccgatggcgtactggcgcactctgttcgacacggacacggtggccggtatttacgatgcgcagacccggaagcagaacggcagcctgagcgaggaggacgcggccctcgtcacggcgcagcaccaggccgccttcgcgacgccgtacctgctgctgcacacgcgcctggtcccgctcttcgggcccgcggtcgagggcccgcccgagatgacggtcgtctttgaccgccacccggtggccgcgacggtgtgcttcccgctggcgcgcttcatcgtcggggacatcagctgcag(seqidno.1);右重组同源臂优选包括部分tk基因和部分ul22基因,大小为976bp,具体序列如下:ctgcagtccaggacaccctcttcggcgcgtacaaggcgcccgagctctgcgaccggcgcgggcgcccgctcgaggtgcacgcgtgggcgatggacgcgctcgtggccaagctgctgccgctgcgcgtctccaccgtcgacctggggccctcgccgcgcgtctgcgccgcggccgtggcggcgcaggcgcgcggcatggaggtgacggagtccgcgtacggcgaccacatccggcagtgcgtgtgcgccttcacgtcggagatgggggtgtgaccctcgcccctcccacccgcgccgcggccggatggagaccgcgacggaggcaacgacgacggcgtgggagggggctcggggcgcgtataaagccatgtgtatgtcatcccaataaagtttgccgtgcccgtcaccatgcccgcgtcgtccgtgcgcctcccgctgcgcctcctgaccctcgcgggcctcctggccctcgcgggggccgccgccctcgcccgcggcgcgccgcagggtgggccgccctcgccgcaggggggtcccgcgcccaccgcggcgcccgcgcgcgggcccaccctgttcgtcctggacggcgacggctccgcgtggttcgtcttccagctcggcgggctgggggcgctcaacgacacgcgcatccgcgggcacctgctcggccggtacctcgtctcgtaccaggtggtgcccccgcccgtctccgcgtggtactttgtgcagcgcccgcgcgagcgcccgcgcctctcggggccgccctcgggcgcggagctcgtggccttcgacgcgcccggcgtctggcgcacgtacaccacggcggcggtgtggcccgcggaggtggccgtcctcgcggacgcggaggcgcgctgccccgcggccgtcttcaacgtgacgctgggcgaggccttcctcggcctgcgcgtcgcgctgcgctccttcctgccgctggaggtcatcatctccgcgaattc(seqidno.2)。本发明对所述左、右重组同源臂的扩增条件和鉴定方法没有特殊的限定,采用常规方法和条件对两段序列进行扩增和鉴定即可。本发明左重组同源臂引物tkl/r的3’端和右重组同源臂引物tkr/f的5’端优选采用相同的酶切位点,用于后续egfp基因表达盒的插入。在本发明中,所述酶切位点优选为pstⅰ。
表1同源臂扩增所需引物信息
注:f表示上游引物,r表示下游引物
在本发明中,所述egfp基因表达盒的核苷酸序列由以下基因片段依次排布组成:cmv、mcs、egfp和sv40polya尾。egfp基因表达盒应用于重组病毒的筛选将会大大提高筛选的正确率,使筛选工作更为便捷。本发明所述egfp基因表达盒的序列大小为1674bp,具体序列如下:ctgcaggattctgtggataaccgtattaccgccatgcattagttattaatagtaatcaattacggggtcattagttcatagcccatatatggagttccgcgttacataacttacggtaaatggcccgcctggctgaccgcccaacgacccccgcccattgacgtcaataatgacgtatgttcccatagtaacgccaatagggactttccattgacgtcaatgggtggagtatttacggtaaactgcccacttggcagtacatcaagtgtatcatatgccaagtacgccccctattgacgtcaatgacggtaaatggcccgcctggcattatgcccagtacatgaccttatgggactttcctacttggcagtacatctacgtattagtcatcgctattaccatggtgatgcggttttggcagtacatcaatgggcgcggatagcggtttgactcacggggatttccaagtctccaccccattgacgtcaatgggagtttgttttggcaccaaaatcaacgggactttccaaaatgtcgtaacaactccgccccattgacgcaaatgggcggtaggcgtgtacggtgggaggtctatataagcagagctggtttagtgaaccgtcagatccgctagcgctaccggactcagatctcgagctcaagcttcgaattcgatccaccggtcgccaccatggtgagcaagggcgaggagctgttcaccggggtggtgcccatcctggtcgagctggacggcgacgtaaacggccacaagttcagcgtgtccggcgagggcgagggcgatgccacctacggcaagctgaccctgaagttcatctgcaccaccggcaagctgcccgtgccctggcccaccctcgtgaccaccttcacctacggcgtgcagtgcttcagccgctaccccgaccacatgaagcagcacgacttcttcaagtccgccatgcccgaaggctacgtccaggagcgcaccatcttcttcaaggacgacggcaactacaagacccgcgccgaggtgaagttcgagggcgacaccctggtgaaccgcatcgagctgaagggcatcgacttcaaggaggacggcaacatcctggggcacaagctggagtacaactacaacagccacaacgtctatatcatggccgacaagcagaagaacggcatcgaggtgaacttcaagatccgccacaacatcgaggacggcagcgtgcagctcgccgaccactaccagcagaacacccccatcggcgacggccccgtgctgctgcccgacaaccactacctgagcacccagtccgccctgagcaaagaccccaacgagaagcgcgatcacatggtcctgctggagttcgtgaccgccgccgggatcactctcggcatggacgagctgtacaagtaaagcggccgcgactctagatcataatcagccataccacatttgtagaggttttacttgctttaaaaaacctcccacacctccccctgaacctgaaacataaaatgaatgcaattgttgttgttaacttgtttattgcagcttataatggttacaaataaagcaatagcatcacaaatttcacaaataaagcatttttttcactgcattctagttgtggtttgtccaaactcatcaatgtatcttaaggcgtaaattgtctgcag(seqidno.3)。在本发明中,所述egfp基因表达盒的来源优选为伪狂犬病病毒基因缺失转移载体(pg),本发明对所述伪狂犬病病毒基因缺失转移载体(pg)的来源没有特殊的限定,优选采用常规报道的伪狂犬病病毒基因缺失转移载体(pg)即可,如朱前磊在《猪圆环病毒ⅱ型orf2和猪il--2基因重组猪伪狂犬病毒的构建》一文中公开的伪狂犬病病毒基因缺失转移载体(pg),本发明所述伪狂犬病病毒基因缺失转移载体(pg)由郑州市猪重大疫病防控重点实验室发明构建。在本发明中,所述egfp作为标记基因,本发明优选采用pstⅰ对pg质粒进行酶切,以便于后续第二转移质粒的构建。本发明所述第一转移质粒和第二转移质粒的构建流程图如图1所示。本发明所述egfp基因表达盒克隆插入的位置在tk基因两侧序列的中间,以便于后续第二次同源重组反向筛选出不含任何外源基因的缺失病毒。得到左右重组同源臂和egfp基因表达盒后,本发明对将所述三段基因克隆至载体上的方法没有特殊的限定,采用本领域技术人员熟知的载体构建方法即可,如酶切连接法。本发明对酶切连接的顺序没有特殊的限定,优选按照先左重组同源臂、然后右重组同源臂、最后egfp基因表达盒的顺序。
本发明将重组猪伪狂犬病毒rprvhn2012-ge-/gi-接种于细胞中,吸附1~2h后,得到经重组猪伪狂犬病毒rprvhn2012-ge-/gi-感染的细胞,将第二转移质粒puc-tklre转染于经重组猪伪狂犬病毒rprvhn2012-ge-/gi-感染的细胞中,通过空斑纯化获得表达荧光蛋白的过渡病毒rprvhn2012-ge-/gi-/tk--egfp+。相比于常规的将病毒吸附和质粒转染同时进行的方法,本发明采用先吸附、后转染的方法,能够大大增加病毒基因组同源重组的效率。在本发明中,接种用细胞优选包括st细胞。在本发明中,所述st细胞待培养至细胞融合70%时,用于接种。本发明对细胞的培养方法、吸附、转染的其它具体操作方法没有特殊的限定,采用本领域技术人员熟知的操作方法即可。在本发明中,所述吸附的时间优选为1.5h。本发明优选采用可生物降解的纳米颗粒转染试剂xfecttmtransfectionreagent进行转染操作。xfect转染试剂是基于可降解的纳米颗粒制备的,对细胞毒性非常低,转染效率高,且在转染过程中兼容血清,无需使用opti-mem培养液,操作简便;在转染过程中,细胞的生长始终处于较为良好的状态,为病毒的重组提供了良好的环境。在本发明中,所述空斑纯化的次数为4~7次,更优选为5次,在蓝光条件下观察到所有病毒蚀斑呈绿色荧光时表明纯化完全。本发明对所述空斑纯化的具体操作条件没有特殊的限定,采用本领域技术人员熟知的常规空斑纯化操作方法即可。
本发明将过渡病毒rprvhn2012-ge-/gi-/tk--egfp+接种于细胞中,吸附1~2h后,得到经过渡病毒rprvhn2012-ge-/gi-/tk--egfp+感染的细胞,将第一转移质粒puc-tklr转染于经过渡病毒rprvhn2012-ge-/gi-/tk--egfp+感染的细胞中,通过空斑纯化去除egfp基因表达盒,得到重组猪伪狂犬病毒rprvhn2012-tk-/ge-/gi-。相比于常规的将病毒吸附和质粒转染同时进行的方法,本发明采用先吸附、后转染的方法,能够大大增加病毒基因组同源重组的效率。在本发明中,接种用细胞优选包括st细胞。在本发明中,所述st细胞待培养至细胞融合70%时,用于接种。本发明对细胞的来源、培养方法、吸附、转染的其它具体操作方法没有特殊的限定,采用本领域技术人员熟知的常规市售st细胞和操作方法即可,本发明所述st细胞优选购自中国兽药监察所。在本发明中,所述吸附的时间优选为1.5h。本发明优选采用可生物降解的纳米颗粒转染试剂xfecttmtransfectionreagent进行转染操作。xfect转染试剂是基于可降解的纳米颗粒制备的,对细胞毒性非常低,转染效率高,且在转染过程中兼容血清,无需使用opti-mem培养液,操作简便;在转染过程中,细胞的生长始终处于较为良好的状态,为病毒的重组提供了良好的环境。在本发明中,所述空斑纯化的次数为4~7次,更优选为5次,在蓝光条件下观察到所有病毒蚀斑不呈现绿色荧光时表明纯化完全。本发明对所述空斑纯化的具体操作条件没有特殊的限定,采用本领域技术人员熟知的常规空斑纯化操作方法即可。
本发明还提供了上述技术方案所述重组猪伪狂犬病毒rprvhn2012-tk-/ge-/gi-或上述技术方案所述制备方法制备得到的重组猪伪狂犬病毒rprvhn2012-tk-/ge-/gi-在制备猪伪狂犬病病毒疫苗中的应用。
下面结合具体实施例对本发明所述的重组猪伪狂犬病毒rprvhn2012-tk-/ge-/gi-及其构建方法和应用做进一步详细的介绍,本发明的技术方案包括但不限于以下实施例。
实施例1
转移质粒puc-tklr以及puc-tklre的构建过程
1方法
1.1病毒增殖培养
将生长状态良好的st细胞接种在底部面积为25cm2的培养瓶中,向其中添加适量含有10%胎牛血清的dmem培养液,随后将其置于37℃、5%co2的条件下培养。待单层st细胞长至培养瓶面积的80%时,用d-hank’s液清洗细胞两次,将prv/hn2012细胞毒株和rprvhn2012-ge-/gi-细胞毒株用不含血清的dmem分别接种至不同的细胞瓶,100μl/瓶,37℃吸附1h后,弃去培养瓶中的吸附液,再用d-hank’s洗两次后,在瓶中加入3ml含有2%血清的dmem作为细胞维持液,置条件为37℃、5%co2的培养箱中培养。待80%细胞发生病变时,将其放入-80℃冰箱中冻融3次收获并保存。
1.2转移质粒构建技术路线图如图1所示。
1.3重组质粒pmd-tkl和pmd-tkr的构建
1.3.1左右重组同源臂的引物设计与合成
参考prvtj株(genbank登录号:kj789182)基因序列,设计两对特异性引物tkl/f和tkl/r、tkr/f和tkr/r(引物信息见表1)分别用于左右重组同源臂的扩增,引物由生工生物工程(上海)股份有限公司合成。其中l同源臂包括部分ul24基因和部分tk基因,预计扩增片段大小为967bp;r同源包括部分tk基因和部分ul22基因,预计扩增片段大小为976bp。
1.3.2左右重组同源臂的扩增
参照生工生物工程(上海)股份有限公司uniq-10柱式病毒基因组抽提试剂盒的说明书对prv/hn2012株基因组进行提取,用于转移质粒puc-tklr左右重组同源臂的pcr扩增。利用两对特异性引物tkl/f和tkl/r、tkr/f和tkr/r,分别扩增tk基因缺失部分两侧的序列作为左右重组同源臂,同源臂上携带各自的酶切位点用于后续质粒的构建(见表1)。
扩增用的pcr反应参数如下:95℃5min;95℃50s;56℃45s;72℃90s共进行35个循环;72℃终延伸10min;4℃10min。反应结束后,取6μlpcr产物用1%琼脂糖凝胶(含0.5mg/leb)进行电泳检测pcr的结果。
1.3.3左右重组同源臂的回收与连接
将1.3.2步骤中扩增的左右重组同源臂使用dna凝胶回收试剂盒进行回收,并对回收的目的片段进行浓度和纯度的测定。
将回收到的特异性片段与pmd18-t载体连接,分别构成pmd-tkl和pmd-tkr重组质粒。连接的加样过程需在冰浴条件下完成,加样完成后将连接产物放置在16℃低温连接槽中过夜即可,所需连接体系如下:dna目的片段5μl,solutioni4μl,pmd18-tvector1μl,共10μl体系。
1.3.4左右重组同源臂的转化与挑斑
分别取5μl连接产物加入50μldh5α感受态细胞中,用于pmd-tkl和pmd-tkr重组质粒的转化。分别随机挑取生长于含氨苄青霉素lb+固体培养平板上的数个白色菌落,各自接种于lb+液体培养基中,37℃条件下振荡过夜,同时设蓝斑为阴性对照。
1.3.5质粒提取、酶切鉴定及序列测定
取1.3.4步骤产物进行菌液pcr鉴定,程序同1.3.2。将pcr检测为阳性的菌液使用质粒小量提取试剂盒提取重组质粒pmd-tkl和pmd-tkr。
将获得的重组质粒pmd-tkl和pmd-tkr通过双酶切进行酶切鉴定,所用体系如下:1μl10×q.cutg.buffer、1μlq.cuthindⅲ、1μlq.cutpstⅰ,2μl重组质粒pmd-tkl或pmd-tkr,加去离子水至10μl体系。
加样完成后置37℃水浴条件下消化30min后,进行电泳观察酶切结果。将菌液pcr和酶切鉴定双阳性的重组质粒送往生工生物工程(上海)股份有限公司测序部进行序列测定,测序正确后通过ncbiblast进行比对,以进一步确定左右重组同源臂克隆的正确性。
1.4转移质粒puc-tklr的构建与鉴定
1.4.1左右重组同源臂的酶切片段回收
将重组质粒pmd-tkl和pmd-tkr分别使用q.cuthindⅲ和q.cutpstⅰ、q.cutecorⅰ和q.cutpstⅰ进行双酶切,体系按照1.3.5中的酶切体系。加样完成后置37℃水浴条件下消化30min。酶切反应结束后,使用dna凝胶回收试剂盒对左右重组同源臂的酶切片段进行胶回收。
1.4.2puc19载体(hindⅲ-pstⅰ)酶切片段回收
将50μl真核表达载体中加入5μl10×q.cutg.buffer、10μlq.cuthindⅲ、10μlq.cutpstⅰ,加去离子水至100μl体系,之后置37℃水浴条件下消化30min。酶切反应结束后,使用dna凝胶回收试剂盒对目的片段进行胶回收。
1.4.3puc-tkl质粒的连接、转化和提取
取酶切回收的左重组同源臂片段7μl、puc19载体(hindⅲ-pstⅰ)片段1μl,再加入10×ligationbuffer1μl、t4dna连接酶1μl共同构成10μl反应体系,加样完成后置16℃连接槽反应过夜,构建质粒puc-tkl。
将连接产物转化并挑斑摇菌,用特异性引物tkl/f和tkl/r对菌液进行菌液pcr鉴定,之后将提取阳性质粒puc-tkl用于后续试验。
1.4.4质粒puc-tkl(ecorⅰ-pstⅰ)酶切片段回收
将50μlpuc-tkl质粒中加入5μl10×q.cutg.buffer、10μlq.cutecorⅰ、10μlq.cutpstⅰ,加去离子水至100μl体系,之后置37℃水浴条件下消化30min。酶切反应结束后,使用dna凝胶回收试剂盒对目的片段进行胶回收。
1.4.5转移质粒puc-tklr的连接、转化和提取
取酶切回收的右重组同源臂片段7μl、质粒puc-tkl(ecorⅰ-pstⅰ)酶切片段1μl,再加入10×ligationbuffer1μl、t4dna连接酶1μl共同构成10μl反应体系,加样完成后置16℃连接槽反应过夜,构建转移质粒puc-tklr。
将连接产物转化并挑斑摇菌,用特异性引物tkr/f和tkr/r对菌液进行菌液pcr鉴定。将鉴定出的阳性菌液使用omegae.z.n.a.tmendo-freeplasmidminikitⅰd6948-01b试剂盒提取转移质粒puc-tklr。将获得的转移质粒puc-tklr用hindⅲ、pstⅰ进行酶切鉴定后(酶切体系参照1.3.5),保存在-20℃条件下以供后续实验使用。
1.5转移质粒puc-tklre的构建与鉴定
1.5.1pg质粒中含egfp基因的(pstⅰ-pstⅰ)酶切片段回收
pg质粒为本发明构建的含绿色荧光蛋白基因的伪狂犬病病毒基因缺失转移质粒(结果见图2),其中(pstⅰ-pstⅰ)部分含有完整的egfp基因表达盒,具体包括cmv、mcs、egfp、sv40polya尾基因片段,近2000bp大小。
将50μlpg质粒中加入5μl10×q.cutg.buffer、10μlq.cutpstⅰ,加去离子水至100μl体系,之后置37℃水浴条件下消化30min。酶切反应结束后,使用dna凝胶回收试剂盒对含egfp基因的目的片段进行胶回收。
1.5.2转移质粒puc-tklr(pstⅰ-pstⅰ)酶切片段回收
将50μlpuc-tklr质粒中加入5μl10×q.cutg.buffer、10μlq.cutpstⅰ,加去离子水至100μl体系,之后置37℃水浴条件下消化30min。酶切反应结束后,使用dna凝胶回收试剂盒对目的片段进行胶回收。
1.5.3转移质粒puc-tklre的连接、转化和提取
取回收的含egfp基因的(pstⅰ-pstⅰ)酶切片段7μl、质粒puc-tklr(pstⅰ-pstⅰ)酶切片段1μl,再加入10×ligationbuffer1μl、t4dna连接酶1μl共同构成10μl反应体系,加样完成后置16℃连接槽反应过夜,构建转移质粒puc-tklre。
将连接产物转化并挑斑摇菌,参照genbankno.cvu55762序列,设计特异性引物egfp/f和egfp/r(egfp/f序列为5’-cagtgcttcagccgctaccc-3’(seqidno.8);egfp/r序列为5’-agttcaccttgatgccgttc-3’(seqidno.9))对菌液进行菌液pcr鉴定,扩增的目的条带长度为289bp,pcr反应参数为:95℃5min;95℃45s;53℃45s;72℃1min,共进行35个循环;72℃终延伸10min;4℃10min。之后使用omegae.z.n.a.tmendo-freeplasmidminikitⅰd6948-01b试剂盒提取阳性质粒puc-tklre用于后续试验,并用pstⅰ对其进行酶切鉴定。
2结果与分析
2.1重组质粒pmd-tkl和pmd-tkr的构建
2.1.1左右重组同源臂的pcr扩增结果
用合成的两对特异性引物tkl/f和tkl/r、tkr/f和tkr/r分别对左右重组同源臂进行pcr扩增,结果经电泳后显示分别扩增出了一条近1000bp大小的片段(同源臂的扩增截图如图3所示,其中,m1.dnamarkerdl2000+;1.左重组同源臂(tkl);2.右重组同源臂(tkr);3.阴性对照),符合预期结果。
2.1.2重组质粒的双酶切结果
用hindⅲ和pstⅰ对pmd-tkl质粒进行酶切,经电泳后出现了2条带,一条为插入的左重组同源臂条带,位置约为1000bp;另外一条为pmd-18t载体带,位置约为2.7kb;用ecorⅰ和pstⅰ对pmd-tkr质粒进行酶切,经电泳后出现了2条带,1条为插入的右重组同源臂条带,位置约为1000bp,另外1条为pmd-18t载体带,位置约为2.7kb(重组质粒pmd-tkl、pmd-tkr的酶切鉴定图如图4所示,其中,m1.dnamarkerdl5000;1.pmd-tkl的hindⅲ/pstⅰ酶切;2.pmd-tkr的ecorⅰ/pstⅰ酶切)。
2.1.3左右重组同源臂的序列测定
将菌液pcr和酶切鉴定双阳性的重组质粒送往生工生物工程(上海)股份有限公司测序部进行序列测定,测序正确后通过ncbiblast进行比对,左、右重组同源臂与prvtj株相应基因区段的同源性均可高达99.9%。
2.2转移质粒puc-tklr的酶切鉴定
将获得的转移质粒puc-tklr用hindⅲ、pstⅰ进行酶切鉴定,电泳结果显示出现了2条带,一条为插入的左重组同源臂,位置约为1000bp;另外一条为右重组同源臂与puc-19载体通过ecorⅰ位点相连的片段,位置约为3700bp;结果与预期相符(转移质粒的酶切鉴定图如图5所示,其中,m1.dnamarkerdl5000;1.puc-tklr的hindⅲ/pstⅰ酶切;2.puc-tklre的pstⅰ酶切)。
2.3转移质粒puc-tklre的构建与鉴定
2.3.1质粒pg的酶切结果
将pg质粒用pstⅰ进行酶切鉴定,电泳结果显示出现了2条带,一条为含egfp基因(pstⅰ-pstⅰ)片段,位置约为1700bp;另外一条为载体片段,位置约为5400bp;结果与预期相符(pg质粒的酶切鉴定图如图6所示,其中,m1.dnamarkerdl5000;1.pg的pstⅰ酶切)。
2.3.2菌液pcr检测结果
用合成的特异性引物egfp/f和egfp/r对含有重组质粒puc-tklre的菌液进行菌液pcr鉴定,阳性菌液经pcr扩增后经电泳跑胶可显示扩增出一条约300bp大小的片段(菌液pcr鉴定图如图7所示,其中,m1.dnamarkerdl2000+;1-5.egfp基因片段;6.阴性对照),符合预期结果。
2.3.3转移质粒puc-tklre的酶切鉴定
将获得的转移质粒puc-tklre用pstⅰ进行酶切鉴定,电泳结果显示出现了2条带,一条为插入的含egfp基因的(pstⅰ-pstⅰ)酶切片段,位置约为1700bp;另外一条为左、右重组同源臂与puc-19载体分别通过hindⅲ、bamhⅰ位点相连的片段,位置约为5200bp;结果与预期相符(如图5)。
本发明以rprvhn2012-ge-/gi-株作为亲本株,将egfp基因作为筛选基因,设计了转移质粒puc-tklr和puc-tklre以实现tk基因的缺失。其中l同源臂包括部分ul24基因和部分tk基因,预计扩增片段大小为967bp;r同源包括部分tk基因和部分ul22基因,预计扩增片段大小为976bp;合适长度的同源臂是保证同源重组顺利进行的基础,转移质粒左右重组同源臂长度完全可以满足需要。
实施例2
重组猪伪狂犬病毒rprvhn2012-tk-/ge-/gi-的拯救与纯化
1rprvhn2012-tk-/ge-/gi-三基因缺失株的构建方法
1.1过渡病毒rprvhn2012-ge-/gi-/tk--egfp+的构建
1.1.1转染细胞的准备
将胰酶消化好的形态良好、生长活力强的st细胞用含有10%胎牛血清的dmem培养液吹打分散成单个细胞,并接种于6孔细胞培养板上,每孔2ml细胞混合液。将该6孔板封口后置于37℃、5%co2细胞培养箱中过夜培养。
1.1.2转染
待细胞融合70%时吸弃原有的培养基,用d-hank’s液清洗细胞两次,换为不含胎牛血清的dmem培养液,将rprvhn2012-ge-/gi-株的病毒液分别接种于细胞培养板中,接毒后的六孔细胞培养板置于37℃、5%co2细胞培养箱中2h,使病毒充分吸附到细胞表面。然后弃去细胞病毒液,用无血清、无抗生素的dmem培养基清洗细胞两次,再换为含有10%胎牛血清的dmem培养液。参照宝生物工程(大连)有限公司的xfecttmtransfectionreagent试剂说明书,将提取的转移质粒puc-tklre转染于rprvhn2012-ge-/gi-株感染过的st细胞中,质粒puc-tklre与rprvhn2012-ge-/gi-基因组发生同源重组,获得重组病毒rprvhn2012-ge-/gi-/tk--egfp+。将六孔板放置37℃、5%co2细胞培养箱中培养4h后,移除含有转染试剂的培养液,换为2ml新鲜的含有10%胎牛血清的dmem培养液,放置培养箱继续培养。约48h后可冻融收取转染液,3000rpm离心5min后取上清至-80℃保存。
1.1.3重组病毒rprvhn2012-ge-/gi-/tk--egfp+的空斑纯化
用无血清和无双抗的dmem细胞培养液对收获的上清病毒液进行10倍的倍比稀释,将稀释好的10-2-10-7梯度的病毒液各取100μl分别接种在单层st细胞密度达到90%的六孔细胞培养板的各个孔中。将接种病毒的六孔板放置于37℃、5%co2细胞培养箱中培养2h。将融化的1.3%低熔点琼脂糖与含4%胎牛血清的2×dmem等比例混合;吸弃吸附结束的含病毒液的培养基,用d-hank’s液清洗细胞两次,每孔覆盖2ml混合好的低熔点琼脂糖,待其完全凝固后移至于37℃、5%co2细胞培养箱中继续培养。每间隔12h在荧光倒置显微镜蓝光下(波长介于420~490nm)观察并记录每个孔中的绿色荧光蚀斑产生情况。大约培养48h后,将最大稀释倍数孔中的最大绿色荧光蚀斑在蓝光条件下进行标记,标记完成后移至无菌操作台内,用移液枪挑取标记好的蚀斑置于含有200μl无血清、无双抗dmem细胞培养液的无菌eppendorf管中并将其捣碎,在常温与-80℃反复冻融三次,每次冻融期间对其进行旋涡振荡。收获的混合液接种st细胞进行增殖,将可在蓝光条件下观察到绿色荧光的阳性病毒液收获重复以上方法进入下一轮蚀斑纯化。纯化过程重复至在蓝光条件下观察到的所有病毒蚀斑均可呈现绿色荧光(大约需进行5次蚀斑纯化),则表明已纯化完全,将得到的病毒命名为rprvhn2012-ge-/gi-/tk--egfp+。
1.1.4重组病毒rprvhn2012-ge-/gi--egfp+的pcr鉴定
参照生工生物工程(上海)股份有限公司uniq-10柱式病毒基因组抽提试剂盒的说明书对rprvhn2012-ge-/gi-/tk--egfp+基因组进行提取。针对本试验中左右同源臂之间的tk基因缺失部分,并参考prvtj株(genbank登录号:kj789182)序列设计了一对特异性引物tkqs/f和tkqs/r(tkqs/f序列为5’-tcgtcggggacatcagc-3’(seqidno.10);tkqs/r序列为5’-gacgggcacggcaaact-3’(seqidno.11))用于重组病毒rprvhn2012-ge-/gi-/tk--egfp+的pcr鉴定,预计扩增的目的条带长度约为2200bp,pcr反应参数为:95℃5min;95℃50s;59℃45s;72℃150s共进行35个循环;72℃终延伸10min;4℃10min。
1.2rprvhn2012-tk-/ge-/gi-三基因缺失病毒株的构建方法
1.2.1转染细胞的准备
将胰酶消化好的形态良好、生长活力强的st细胞用含有10%胎牛血清的dmem培养液吹打分散成单个细胞,并接种于6孔细胞培养板上,每孔2ml细胞混合液。将该6孔板封口后置于37℃、5%co2细胞培养箱中过夜培养。
1.2.2转染
待细胞融合70%时吸弃原有的培养基,用d-hank’s液清洗细胞两次,换为不含胎牛血清的dmem培养液,将rprvhn2012-ge-/gi-/tk--egfp+株的病毒液接种于细胞培养板中,接毒后的六孔细胞培养板置于37℃、5%co2细胞培养箱中2h,使病毒充分吸附到细胞表面。然后弃去细胞病毒液,用无血清、无抗生素的dmem培养基清洗细胞两次,再换为含有10%胎牛血清的dmem培养液。参照宝生物工程(大连)有限公司的xfecttmtransfectionreagent试剂说明书,将提取的转移质粒puc-tklr转染于rprvhn2012-ge-/gi-/tk--egfp+株感染过的st细胞中,将提取的转移质粒puc-tklre转染于rprvhn2012-ge-/gi-/tk--egfp+株感染过的st细胞中,质粒puc-tklr与rprvhn2012-ge-/gi-/tk--egfp+基因组发生同源重组,获得重组病毒rprvhn2012-tk-/ge-/gi-。将六孔板放置37℃、5%co2细胞培养箱中培养4h后,移除含有转染试剂的培养液,换为2ml新鲜的含有10%胎牛血清的dmem培养液,放置培养箱继续培养。约48h后可冻融收取转染液,3000rpm离心5min后取上清至-80℃保存。
1.2.3重组病毒rprvhn2012-tk-/ge-/gi-的空斑纯化
按照1.1.3的步骤,对获取的转染液进行稀释铺胶以完成rprvhn2012-tk-/ge-/gi-的纯化。每次蚀斑纯化注意观察病毒蚀斑形态,挑选最大稀释倍数孔中的在荧光倒置显微镜蓝光条件下看不到绿色荧光的蚀斑。纯化过程重复至在蓝光条件下观察到的所有病毒蚀斑均不呈现绿色荧光(大约需进行5次蚀斑纯化),则表明已纯化完全,将得到的病毒命名为rprvhn2012-tk-/ge-/gi-。
1.2.4重组病毒rprvhn2012-tk-/ge-/gi-的pcr鉴定
参照生工生物工程(上海)股份有限公司uniq-10柱式病毒基因组抽提试剂盒的说明书对重组病毒rprvhn2012-tk-/ge-/gi-基因组进行提取。使用1.1.4中设计的特异性引物tkqs/f和tkqs/r,对重组病毒rprvhn2012-tk-/ge-/gi-进行缺失部分的鉴定,pcr反应参数为参照1.1.4。对pcr扩增出的目的条带进行回收、连接、转化以及挑斑,之后将阳性菌液送至生工生物工程(上海)股份有限公司测序部进行测序,以进一步确定是否成功对rprvhn2012-ge-/gi-株进行了tk基因的进一步缺失。之后参考prvtj株(genbank登录号:kj789182)设计的一对gb全基因引物(gb/f序列为5’-ttgcagtcttcaggtcggtctt-3’(seqidno.12);gb/r序列为5’-tattatctgcggggagggggct-3’(seqidno.13))对rprvhn2012-tk-/ge-/gi-进行gb基因的检测。
1.2.5重组病毒rprvhn2012-tk-/ge-/gi-的生长特性
将缺失病毒rprvhn2012-tk-/ge-/gi-和rprvhn2012-ge-/gi-、弱毒疫苗bartha-k61株以及亲本病毒prvhn2012株分别以10μl(106.0tcid50/0.1ml)剂量接种24孔板中单层的st细胞,37℃、5%co2条件下吸附2h后弃去病毒液,更换成含2%胎牛血清的dmem维持液进行培养。在接种病毒4h后开始每隔2h收取细胞及其上清培养液,并于-80℃条件下反复冻融3次后进行tcid50值的测定,根据结果绘制出病毒的一步生长曲线,并进行分析。
2结果与分析
2.1rprvhn2012-ge-/gi-/tk--egfp+三基因缺失株的构建
2.1.1rprvhn2012-ge-/gi-/tk--egfp+的拯救结果
rprvhn2012-ge-/gi-双基因缺失病毒基因组与转移质粒puc-tklre进行共转染48h后收获转染液,冻融该转染产物后经10倍系列稀释进行蚀斑筛选。第一次纯化时每孔内均可观察到表达绿色荧光的蚀斑和不表达绿色荧光的蚀斑,且不表达绿色荧光的蚀斑多于表达绿色荧光的蚀斑。每次纯化时挑取最大稀释倍数孔中的最大绿色荧光蚀斑(不同光照下观察到的蚀斑照片如图8所示,其中,图8a:常光下rprvhn2012-ge-/gi-/tk--egfp+形成的蚀斑;图8b为蓝光下rprvhn2012-ge-/gi-/tk--egfp+形成的蚀斑),随着纯化次数的增加绿色荧光蚀斑逐渐增加,直至第五次纯化时观察到的蚀斑均可表达绿色荧光,即成功拯救并纯化获得了可表达绿色荧光的三基因缺失株rprvhn2012-ge-/gi-/tk--egfp+。
2.1.2重组病毒rprvhn2012-ge-/gi-/tk--egfp+的pcr鉴定结果
提取rprvhn2012-ge-/gi-/tk--egfp+和rprvhn2012-ge-/gi-基因组,通过特异性引物tkqs/f和tkqs/r对其左右同源臂见的缺失部分进行鉴定,结果显示rprvhn2012-ge-/gi-/tk--egfp+基因组扩增出了一条约2200bp大小的片段,未缺失tk基因的rprvhn2012-ge-/gi-基因组扩增出了一条约725bp大小的片段(tk缺失部分pcr鉴定结果如图9所示,其中,m1.dnamarkerdl5000;1.rprvhn2012-ge-/gi-的缺失鉴定结果;2.rprvhn2012-ge-/gi-/tk--egfp+的缺失鉴定结果),与预期结果相符。
2.2rprvhn2012-tk-/ge-/gi-三基因缺失株的构建
2.2.1rprvhn2012-tk-/ge-/gi-的拯救结果
rprvhn2012-ge-/gi-/tk--egfp+基因组与转移质粒puc-tklr进行共转染48h后收获转染液,对10倍稀释的转染产物进行蚀斑筛选。在筛选过程中观察到可产生细胞病变但并不表达绿色荧光的蚀斑(不同光照下观察到的蚀斑照片如图10所示,其中,图10a:常光下rprvhn2012-tk-/ge-/gi-形成的蚀斑;图10b:蓝光下rprvhn2012-tk-/ge-/gi-形成的蚀斑),可以初步认为是去除egfp基因的重组病毒rprvhn2012-tk-/ge-/gi-。每次挑选最大稀释倍数孔中的在荧光倒置显微镜蓝光条件下看不到绿色荧光的蚀斑,随着纯化次数的增加不表达绿色荧光蚀斑逐渐增加,直至第五次纯化时观察到的蚀斑均不表达绿色荧光,即成功拯救并纯化获得了去除egfp基因的三基因缺失株rprvhn2012-tk-/ge-/gi-。
2.2.2重组病毒rprvhn2012-tk-/ge-/gi-的pcr鉴定结果
提取rprvhn2012-tk-/ge-/gi-基因组,通过特异性引物tkqs/f和tkqs/r对其左右同源臂间的缺失部分进行鉴定,结果显示rprvhn2012-ge-/gi-/tk-/egfp+株扩增出了一条约2200bp大小的片段,而rprvhn2012-tk-/ge-/gi-株扩增出了一条约420bp大小的片段(tk缺失部分pcr鉴定结果如图11所示,其中,m1.dnamarkerdl5000+;1.rprvhn2012-ge-/gi-/tk-/egfp+的缺失鉴定结果;2.rprvhn2012-tk-/ge-/gi-的缺失鉴定结果),与预期结果相符。
测序结果显示,rprvhn2012-tk-/ge-/gi-三基因缺失株成功缺失了311bp的tk基因片段,并在缺失部位留下了一个pstⅰ位点,符合预期结果。
2.2.3重组病毒rprvhn2012-tk-/ge-/gi-的gb基因鉴定结果
通过gb全基因引物对重组病毒rprvhn2012-tk-/ge-/gi-的gb基因进行鉴定,结果显示扩增出了约3000bp大小的片段(重组病毒rprvhn2012-tk-/ge-/gi-gb基因的鉴定结果如图12所示,其中,m1.dnamarkerdl5000;1-2.rprvhn2012-tk-/ge-/gi-的gb基因鉴定结果;3.阴性对照),与预期结果相符。
2.2.4重组病毒rprvhn2012-tk-/ge-/gi-的生长特性结果
缺失病毒和亲本毒株的一步生长曲线图如图13所示,根据测定结果绘制处图13所示的一步生长曲线,可以看出:三基因缺失毒株rprvhn2012-tk-/ge-/gi-、双基因缺失毒株rprvhn2012-ge-/gi-、以及弱毒疫苗bartha-k61株具有与亲本病毒prv/hn2012株的生长动力学非常相似。在感染细胞后10~14h期间,亲本病毒prv/hn2012株的增殖速度极快,而三基因缺失毒株rprvhn2012-tk-/ge-/gi-、双基因缺失毒株rprvhn2012-ge-/gi-和弱毒疫苗bartha-k61株的增值速度和病毒滴度都低于亲本株。另外,三基因缺失毒株rprvhn2012-tk-/ge-/gi-、双基因缺失毒株rprvhn2012-ge-/gi-、以及弱毒疫苗bartha-k61株接种st细胞后形成的蚀斑在形态和大小上与亲本病毒prv/hn2012株上并未有明显差异。
3结论与讨论
通过转移质粒puc-tklre和puc-tklr分别与prv基因组的两次同源重组,先获得表达绿色荧光蛋白的rprvhn2012-ge-/gi-/tk--egfp+三基因缺失株,再通过反向筛选获得了不含外源egfp基因的rprvhn2012-tk-/ge-/gi-三基因缺失株。进一步结合pcr鉴定及测序,我们获得的rprvhn2012-tk-/ge-/gi-三基因缺失株成功缺失了311bp片段,并在缺失部位留下了一个pstⅰ位点。
本发明进行两次转染,通过转移质粒puc-tklr与rprvhn2012-ge-/gi-/tk--egfp+基因组的同源重组,经反向筛选方法挑取不含荧光蛋白的蚀斑,并结合pcr鉴定技术,成功获得了不含外源egfp基因的rprvhn2012-tk-/ge-/gi-三基因缺失株,从而大大提高了本缺失病毒的稳定性和生物安全性。
进行转染时良好的细胞状态、合适的转染试剂及转移质粒的构建都将直接影响到病毒重组效率的高低。
实施例3
三基因缺失病毒免疫效力初步探究
1方法
1.1小鼠免疫
将30只生长健康的6周龄spf雌性小鼠随机分为3组,每组10只,试验具体分组和接种情况见表2,接种途径为背部皮下分点注射。第一次免疫之前随机在小鼠中选取20只进行尾静脉采血,采血后按分组分别进行免疫;首免两周后对小鼠进行第二次免疫。一免之后每周对小鼠进行尾静脉采血,每次采血后分离血清保存至-20℃用于后续实验,采血至一免后第6周。免疫后每天观察并记录小鼠临床反应情况。
表2试验鼠分组及免疫情况
1.2间接免疫吸附测定(elisa)
本发明参照折尕才措的研究建立了猪伪狂犬病病毒间接elisa检测方法,对每周分离的小鼠血清进行prv抗体水平检测,具体方法如下:
(1)用包被液对纯化的prv/hn2012株抗原进行包被(抗原的最佳包被量为0.8μg/ml),酶标板中每孔加100μl,将其置于4℃条件下过夜(包被时间应大于12h)。
(2)过夜后将板子中液体甩净,在吸水纸上拍干,并用pbst溶液将酶标板清洗三次。
(3)每孔中加270μl1%bsa封闭液,置于37℃条件下进行2h封闭。
(4)封闭结束后将板子中液体甩净,在吸水纸上拍干,并用pbst溶液将酶标板清洗一次。
(5)将阴性血清、阳性血清以及每组的待检血清用pbs溶液等量混合,每个稀释度重复2个孔,每孔加100μl,每个样品重复2个孔,每孔加100μl。
(6)作用结束后将板子中液体甩净,在吸水纸上拍干,并用pbst溶液将酶标板清洗四次。
(7)用pbs将羊抗鼠辣根过氧化物酶标记的二抗按1:5000稀释,每孔加入100μl,将酶标板置于37℃条件下1h。
(8)孵育结束后将板子中液体甩净,在吸水纸上拍干,并用pbst溶液将酶标板清洗四次。
(9)每空加入100μltmb,37℃条件下避光显色10min。
(10)避光显色结束后,每孔加入50μl终止液以终止反应。
(11)在反应终止的3min内,置酶标仪450nm波长处测定隔空od值,记录后分析结果。
(12)判定标准:本试验建立的elisa方法阴阳性临界值为0.382,即样品od450>0.382判为阳性,od450<0.382判为阴性。
1.3血清中和试验检测prv抗体水平
用病毒中和试验(终点法)固定病毒-稀释血清法(β法)对每周采集的小鼠血清测定中和抗体效价,具体操作如下:
(1)将试验中所用血清放置在56℃条件进行30min水浴灭活。
(2)用不含血清的dmem培养液将灭活后的血清进行2倍系列稀释,每个稀释度取50μl加入96孔板孔中,每个稀释度设置四个重复。
(3)将50μl含200个tcid50/0.1ml的prv/hn2012株病毒液加置上述各个孔中,这样与孔中50μl待检血清充分混合后,每孔中含有100个tcid50的病毒液。
(4)病毒液与待检血清充分混合后置于37℃条件下作用1h;1h后将混合液接种到铺有单层st细胞的96孔板中。
(5)作用1h后,移除板中混合液,每孔加入200μl含2%胎牛血清的dmem细胞培养维持液,放置在37℃、5%co2环境中连续培养四天,每天观察记录细胞病变情况,用karber计算方法对结果进行判定。
(6)试验中还应设置以下对照组:
①病毒对照:需将0.2、2、20和200tcid50/0.1ml浓度的病毒液与等量最低稀释度的待检血清充分混合(混合后每个接种孔病毒浓度为0.1、1、10和100tcid50),感作之后移至铺有单层st细胞的96孔板中;判定结果时,0.1tcid50病毒液孔中应未发生细胞病变,100tcid50应一定会发生细胞病变。
②血清毒性对照:设置一组在组织培养细胞中仅添加被检血清的最低稀释度的对照组,用于检测血清对组织细胞无毒性作用。
③宿主对照:每个反应板中应设置一组仅含组织细胞的空白对照组,组织细胞在整个试验中应始终保持健康状况。
④阳性和阴性血清对照:阳性血清需呈现应有的中和抗体效价,阴性血清没有中和抗体效价。
以上各个对照组成立的条件下,才可以进行待检血清中和抗体效价的判定。
1.4小鼠外周血t淋巴细胞亚群的测定
首免后第6周(即二免后第4周)随机在每组抽取5只小鼠,每只小鼠采集300μl的抗凝血,进行外周血t淋巴细胞亚群的测定,具体操作步骤如下:
(1)每份血清中分别加入1μl荧光标记的抗cd3+、cd4+、cd8+单克隆抗体,轻柔地充分混匀后,在室温条件下孵育30min。
(2)孵育结束后,每个血液样品中加入2ml溶血素,轻柔地混合均匀后,在室温条件下孵育20min。
(3)孵育结束后2000rpm离心5min,之后再加入2ml2%fbs-pbs,继续再以2000rpm离心5min。
(4)最后加入400μlpbs缓冲液定容,使用流式细胞仪对其进行cd3+、cd4+、cd8+t细胞数量的检测。
1.5攻毒保护试验
一免后第六周(二免后第4周)从每组中随机抽取5只小鼠,对每只小鼠以背部皮下注射方式prv/hn2012株的攻毒,攻毒剂量为每只小鼠500μl(病毒滴度为106tcid50/0.1ml),之后连续饲养4周进行观察,记录并分析小鼠的免疫保护情况,同时绘制生存曲线。
1.6脑部病理切片
取各组发病死亡的小鼠和c组未死亡的小鼠脑部,石蜡包埋后切片,观察病理变化。
2结果与分析
2.1安全性
小鼠进行免疫后,dmem(a组)、bartha-k61(b组)和rprvhn2012-tk-/ge-/gi-(c组)免疫组小鼠均未出现不良症状,全部存活。
2.2间接elisa检测prv抗体结果
应用本发明建立的猪伪狂犬病病毒间接elisa检测方法对每周采集的各组小鼠血清进行prv抗体检测,所测得的数据使用graphpadprism6.0绘制成折线图并计算p值,小鼠ppv抗体检测结果如图14所示。从图中我们看出,第一次免疫7d后bartha-k61(b组)和rprvhn2012-tk-/ge-/gi-(c组)已有效地刺激小鼠产生了抗prv的抗体(p<0.001);之后bartha-k61免疫组(b组)和rprvhn2012-tk-/ge-/gi-免疫组(c组)抗体水平均保持稳定升高,在整个试验过程rprvhn2012-tk-/ge-/gi-免疫组(c组)抗体水平略高于bartha-k61免疫组(b组),但两组抗体水平差异并不十分明显(p>0.05)。试验中的dmem免疫组并未刺激小鼠产生prv抗体。
2.3血清中和试验检测prv抗体结果
进行病毒中和试验对每周采集的各组小鼠血清进行prv中和抗体检测,所测得的数据使用graphpadprism6.0绘制成折线图,prv中和抗体效价水平(log2)结果如图15所示。从图中我们看出,第一次免疫7d后bartha-k61(b组)和rprvhn2012-tk-/ge-/gi-(c组)已有效地刺激小鼠产生了抗prv的抗体,但抗体滴度较低;首免后21d(二免后第一周)时bartha-k61(b组)和rprvhn2012-tk-/ge-/gi-(c组)的抗体水平都得到了显著提高,并持续增长。试验结果说明bartha-k61(b组)和rprvhn2012-tk-/ge-/gi-(c组)在刺激小鼠机体产生prv中和抗体水平方面均有明显作用,且dmem免疫组并未刺激小鼠产生prv中和抗体。
2.4小鼠外周血t淋巴细胞亚群的测定
通过流式细胞技术检测小鼠外周血t淋巴细胞亚群的分布情况,所测得的数据使用graphpadprism6.0绘制成柱状图并计算p值,小鼠外周血中cd3+、cd4+、cd8+t淋巴细胞数量的测定结果见图16(*:p<0.05、**:p<0.01、***:p<0.001)。从图中我们可以观察到与dmem免疫的对照组(a组)相比,bartha-k61(b组)和rprvhn2012-tk-/ge-/gi-(c组)免疫的试验组小鼠的外周血t淋巴细胞亚类cd3+、cd4+和cd8+的数量较多,且rprvhn2012-tk-/ge-/gi-(c组)免疫组的t淋巴细胞亚类cd3+、cd4+和cd8+的数量最多。
2.5攻毒保护试验
dmem免疫组(a组)在攻prv/hn2012株48h后有两只小鼠最先开始出现发痒抓挠等典型的pr发病症状,随后该组另外三只小鼠陆续开始发病,攻毒72h内dmem免疫组(a组)小鼠全部死亡,小鼠从发病到死亡经历的时间短、发病迅速。bartha-k61免疫组(b组)在攻prv/hn2012株48h后有一只小鼠开始出现发痒抓挠等典型的pr发病症状,攻毒60h、72h后又各出现一只小鼠开始发病,三只发病小鼠均经历2~3d的病程后死亡;其他两只小鼠在4周观察期内均健康成长,保护率为40%。rprvhn2012-tk-/ge-/gi-免疫组(c组)在攻prvhn2012株48h后有一只小鼠最先开始出现发痒抓挠等典型的pr发病症状,攻毒72h后又出现一只小鼠开始发病,两只发病小鼠也是在经历2~3d的病程后死亡;该组存活3只小鼠,保护率为60%。根据每组小鼠的死亡情况,绘制了如图17所示的小鼠的生存曲线图。
死亡小鼠的伤口状态结果如图18所示。dmem免疫组(a组)发病小鼠死亡迅速,皮肤仅有少量破损处(见图18a);而bartha-k61免疫组(b组)和rprvhn2012-tk-/ge-/gi-免疫组(c组)中的发病小鼠因经历2~3d的病程后才出现死亡,所以小鼠发痒抓挠皮肤出血十分严重,产生了大片损伤(见图18b)。
2.6脑部病理切片
观察病理切片显示,dmem免疫组(a组)死亡小鼠的脑组织中血管周围可见小胶质细胞聚集(图19),炎症反应明显,小鼠的脑部病理切片结果如图20所示,同时脑神经细胞内可见大量核内嗜酸性包涵体(图20a);bartha-k61免疫组(b组)和rprvhn2012-tk-/ge-/gi-免疫组(c组)死亡小鼠的脑神经细胞内可见少量核内嗜酸性包涵体(图20b和图20c),而c组存活小鼠的脑神经细胞内未见核内嗜酸性包涵体(图20d)。
3结论与讨论
实施例3中我们在小鼠模型上对获得的rprvhn2012-tk-/ge-/gi-缺失病毒进行了安全性和免疫效力的初步评价。rprvhn2012-tk-/ge-/gi-免疫组小鼠在试验过程中没有出现任何不良反应,病毒致弱情况理想。间接elisa结果表明rprvhn2012-tk-/ge-/gi-免疫组在免疫过程中产生了较高滴度的prv抗体,中和抗体水平也证实了该缺失病毒保持了良好的免疫原性,此外小鼠外周血t淋巴细胞亚群的测定结果也表明该缺失病毒有效地刺激小鼠机体产生细胞免疫。小鼠的脑部病理切片也说明了弱毒株在一定程度上也阻碍了强毒株对神经组织的侵袭,发挥了一定的保护作用。
总之,本发明的rprvhn2012-tk-/ge-/gi-三基因缺失病毒对非靶动物小鼠是安全的,免疫后能有效并迅速产生prv特异性抗体,且能有效地激活细胞免疫,是一株有希望应用于当前prv流行防控工作的疫苗株。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
序列表
<110>河南农业大学
<120>重组猪伪狂犬病毒rprvhn2012-tk-/ge-/gi-及其构建方法和应用
<160>13
<170>siposequencelisting1.0
<210>1
<211>967
<212>dna
<213>人工序列(artificialsequence)
<400>1
aagcttgccttatcatccccgctccccgccgccgcccggcccggccccgcgcgcgccgcg60
atcgcgatcaccgccgcggcccggcgacgtactcggcgaggccgcgcacggtcgcggcca120
tcgcgctcgcgttgccgcgcgtctgggtgcagggcaggcgcgtcacgtcgagcacgcgca180
tgctccgctgggccacaaacaccagcaggggcacgagcgtgatctcctcgccgcccgggg240
gcacggcggcggcgaggaggcgcgccgagtcgcgcagctggcacagcccctcgtgccgct300
gcccgcgcttgctgggcgtgttgaggttccgggggaagcggcacgtcttgagctcgatga360
ggaagcacaggtgcgggcccgcccccagccgcaccacgcacacgcagtcggggcggcgca420
ccccgaggttgacttcaaaggccagggtcaaggacgccttcttaagcgtctctcggggaa480
gcccgaagagactctcgccgtacgcggacgggtcgcgtcgcaggcgttcgtagaagcggt540
tgtggcagcggatccccgcccggaagcgcgccgggatgcgcatcctccggatctacctcg600
acggcgcctacggcaccggcaagagcaccacggcccgggtgatggcgctcggcggggcgc660
tgtacgtgcccgagccgatggcgtactggcgcactctgttcgacacggacacggtggccg720
gtatttacgatgcgcagacccggaagcagaacggcagcctgagcgaggaggacgcggccc780
tcgtcacggcgcagcaccaggccgccttcgcgacgccgtacctgctgctgcacacgcgcc840
tggtcccgctcttcgggcccgcggtcgagggcccgcccgagatgacggtcgtctttgacc900
gccacccggtggccgcgacggtgtgcttcccgctggcgcgcttcatcgtcggggacatca960
gctgcag967
<210>2
<211>976
<212>dna
<213>人工序列(artificialsequence)
<400>2
ctgcagtccaggacaccctcttcggcgcgtacaaggcgcccgagctctgcgaccggcgcg60
ggcgcccgctcgaggtgcacgcgtgggcgatggacgcgctcgtggccaagctgctgccgc120
tgcgcgtctccaccgtcgacctggggccctcgccgcgcgtctgcgccgcggccgtggcgg180
cgcaggcgcgcggcatggaggtgacggagtccgcgtacggcgaccacatccggcagtgcg240
tgtgcgccttcacgtcggagatgggggtgtgaccctcgcccctcccacccgcgccgcggc300
cggatggagaccgcgacggaggcaacgacgacggcgtgggagggggctcggggcgcgtat360
aaagccatgtgtatgtcatcccaataaagtttgccgtgcccgtcaccatgcccgcgtcgt420
ccgtgcgcctcccgctgcgcctcctgaccctcgcgggcctcctggccctcgcgggggccg480
ccgccctcgcccgcggcgcgccgcagggtgggccgccctcgccgcaggggggtcccgcgc540
ccaccgcggcgcccgcgcgcgggcccaccctgttcgtcctggacggcgacggctccgcgt600
ggttcgtcttccagctcggcgggctgggggcgctcaacgacacgcgcatccgcgggcacc660
tgctcggccggtacctcgtctcgtaccaggtggtgcccccgcccgtctccgcgtggtact720
ttgtgcagcgcccgcgcgagcgcccgcgcctctcggggccgccctcgggcgcggagctcg780
tggccttcgacgcgcccggcgtctggcgcacgtacaccacggcggcggtgtggcccgcgg840
aggtggccgtcctcgcggacgcggaggcgcgctgccccgcggccgtcttcaacgtgacgc900
tgggcgaggccttcctcggcctgcgcgtcgcgctgcgctccttcctgccgctggaggtca960
tcatctccgcgaattc976
<210>3
<211>1674
<212>dna
<213>人工序列(artificialsequence)
<400>3
ctgcaggattctgtggataaccgtattaccgccatgcattagttattaatagtaatcaat60
tacggggtcattagttcatagcccatatatggagttccgcgttacataacttacggtaaa120
tggcccgcctggctgaccgcccaacgacccccgcccattgacgtcaataatgacgtatgt180
tcccatagtaacgccaatagggactttccattgacgtcaatgggtggagtatttacggta240
aactgcccacttggcagtacatcaagtgtatcatatgccaagtacgccccctattgacgt300
caatgacggtaaatggcccgcctggcattatgcccagtacatgaccttatgggactttcc360
tacttggcagtacatctacgtattagtcatcgctattaccatggtgatgcggttttggca420
gtacatcaatgggcgcggatagcggtttgactcacggggatttccaagtctccaccccat480
tgacgtcaatgggagtttgttttggcaccaaaatcaacgggactttccaaaatgtcgtaa540
caactccgccccattgacgcaaatgggcggtaggcgtgtacggtgggaggtctatataag600
cagagctggtttagtgaaccgtcagatccgctagcgctaccggactcagatctcgagctc660
aagcttcgaattcgatccaccggtcgccaccatggtgagcaagggcgaggagctgttcac720
cggggtggtgcccatcctggtcgagctggacggcgacgtaaacggccacaagttcagcgt780
gtccggcgagggcgagggcgatgccacctacggcaagctgaccctgaagttcatctgcac840
caccggcaagctgcccgtgccctggcccaccctcgtgaccaccttcacctacggcgtgca900
gtgcttcagccgctaccccgaccacatgaagcagcacgacttcttcaagtccgccatgcc960
cgaaggctacgtccaggagcgcaccatcttcttcaaggacgacggcaactacaagacccg1020
cgccgaggtgaagttcgagggcgacaccctggtgaaccgcatcgagctgaagggcatcga1080
cttcaaggaggacggcaacatcctggggcacaagctggagtacaactacaacagccacaa1140
cgtctatatcatggccgacaagcagaagaacggcatcgaggtgaacttcaagatccgcca1200
caacatcgaggacggcagcgtgcagctcgccgaccactaccagcagaacacccccatcgg1260
cgacggccccgtgctgctgcccgacaaccactacctgagcacccagtccgccctgagcaa1320
agaccccaacgagaagcgcgatcacatggtcctgctggagttcgtgaccgccgccgggat1380
cactctcggcatggacgagctgtacaagtaaagcggccgcgactctagatcataatcagc1440
cataccacatttgtagaggttttacttgctttaaaaaacctcccacacctccccctgaac1500
ctgaaacataaaatgaatgcaattgttgttgttaacttgtttattgcagcttataatggt1560
tacaaataaagcaatagcatcacaaatttcacaaataaagcatttttttcactgcattct1620
agttgtggtttgtccaaactcatcaatgtatcttaaggcgtaaattgtctgcag1674
<210>4
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>4
aagcttgccttatcatccccgct23
<210>5
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>5
ctgcagctgatgtccccgacgat23
<210>6
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>6
ctgcagtccaggacaccctcttc23
<210>7
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>7
gaattcgcggagatgatgacctc23
<210>8
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>8
cagtgcttcagccgctaccc20
<210>9
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>9
agttcaccttgatgccgttc20
<210>10
<211>17
<212>dna
<213>人工序列(artificialsequence)
<400>10
tcgtcggggacatcagc17
<210>11
<211>17
<212>dna
<213>人工序列(artificialsequence)
<400>11
gacgggcacggcaaact17
<210>12
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>12
ttgcagtcttcaggtcggtctt22
<210>13
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>13
tattatctgcggggagggggct22
- 还没有人留言评论。精彩留言会获得点赞!