一种LRFFT2细胞的构建方法与流程
本发明属于生物
技术领域:
,具体涉及一种lrfft2细胞的构建方法。
背景技术:
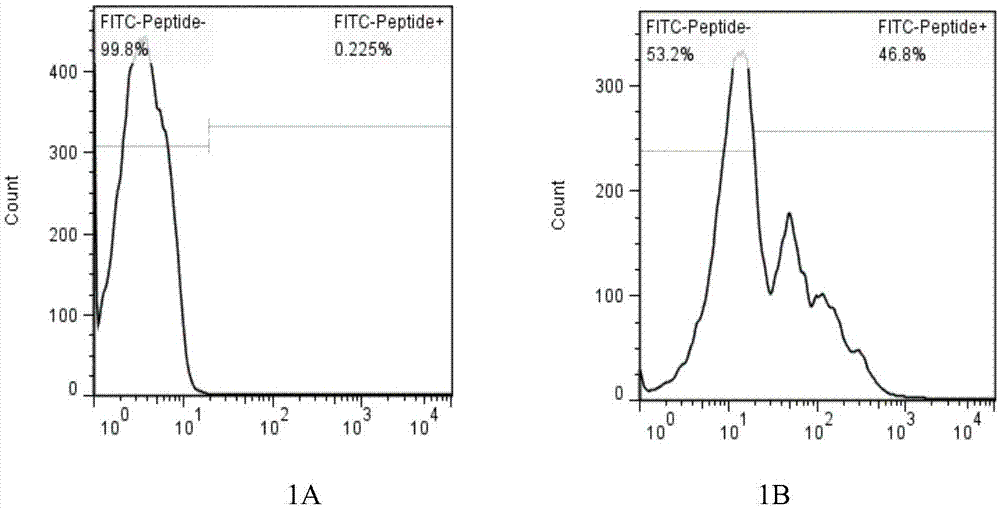
:目前,在肿瘤的特异性免疫治疗方面,现有的lak、dc、cik、dc-cik细胞和方法基本被证明是无效的,而nk、car-nk、til等细胞技术还有待成熟,car-t细胞在安全性和实体瘤治疗中还有缺陷。现有技术一般通过改造dc细胞,由dc递呈t细胞产生特异杀伤。有些实验室在尝试用病毒做为载体的方法进行转染递呈t细胞,诱导t细胞的特异性杀伤。我们也曾用突变混合多肽直接刺激pbmc,诱导t细胞。还有实验室利用tcr-t技术,靶向递呈magea3抗原。以上治疗方法并不成熟,尤其是体外诱导dc细胞及dc细胞负载肿瘤抗原技术理论上研究较多,但在具体实施过程中还有许多问题,缺乏明确的、肿瘤细胞发生发展关键的信号传导通路相关分子作为诱导抗原,因为肿瘤抗原不明及肿瘤微环境免疫抑制的障碍,使实现特异性细胞靶向免疫治疗难以顺利实施。另外,有的虽然进行了抗原体外冲击,但没有进行体外共培育和体外扩增,让较为单薄的特异性细胞直接面对复杂的肿瘤免疫微环境,因此,很难起到预期的效果。也有的虽然也可以体外递呈和共培育,但靶点单一(mage-3),仅对非小细胞肺癌等个别癌种起效。虽然也有尝试慢病毒为载体的方法进行转染递呈,但安全性、方便性不如多肽方式。而简单混合多肽的直接刺激,虽然简单方便,但效率较低。特异性精准多肽的二次刺激不如t细胞受体转导的肿瘤特异性抗原更直接。现有的tcr-t在治疗血液肿瘤和实体肿瘤的解决方案中,缺乏覆盖更多瘤种的精准的tcr。上述方案均没有考虑t细胞的自我防护技术,使得数量不多的特异性t细胞直接面对强大的肿瘤免疫微环境。普遍缺乏对患者抗原也缺乏准确有效的分析。技术实现要素:本发明使用患者外周血进行ctdna测序或肿瘤组织进行全外显子测序,筛选出突变位点进行抗原表位预测,连接并合成突变多肽表达基因序列;同时构建慢病毒载体,包装慢病毒,转染apc细胞,完成特异性lv细胞的改造,体外与从外周血中分离的pbmc共培养,筛选出有效多肽,通过精准有效多肽刺激的第二次冲击,将普通t细胞改造成为具有更精准杀伤能力的rff细胞;再利用tcr-t技术原理进行了改造;改造后的t细胞在体外用抗体药物进行免疫抑制性信号的封闭,精准的保护了特异性杀伤t细胞免受体内抑制,提高了t细胞对肿瘤细胞的杀伤力,得到更为攻防兼备的lrfft2细胞。本发明提供的lrfft2细胞可广泛应用于个体化精准治疗实体肿瘤。对于专用名词的解释:l:慢病毒转染技术r:精准多肽二次冲击技术ff:混合多肽技术t:tcr-t技术2:抗体药物体外封闭防护技术例如:lrfft2细胞,即经由上述l、r、ff、t、2各项技术方案或技术手段改造而最终获得的细胞。lrfft2细胞改造方案概述如下:1、抗原表位预测1)使用人源外周血进行ctdna测序或市售工程细胞系(如h1299、h226、h358、h1563、h2228、a549、renca、llc小鼠lewis肺癌细胞、crl-6323b16f1、crl-25394t1、u14小鼠子宫颈癌细胞、bv-2小鼠小胶质瘤细胞、g422小鼠神经胶质瘤细胞等)进行mhc类型检测和全外显子测序检测rna突变;2)利用mhc类型和基因突变信息预测抗原表位:以突变的氨基酸位点为中心,向两侧延伸8个氨基酸,将这段17个氨基酸的多肽作为潜在抗原表位;3)使用预测软件分析潜在抗原表位的ic50,如ic50<1000nm则认为此潜在抗原表位为抗原表位;2、多肽连接1)使用前述软件分析任意抗原表位两两相连后接头处的ic50,ic50≥1000nm时认为是弱免疫原性,可以连接;ic50<1000nm时认为是强免疫原性,不能连接;2)根据上述结果,将弱免疫原性的抗原表位连接在一起,接头处ic50要高于两侧抗原表位的ic50(也就是接头处尽量避免产生强结合抗原);3、合成编码多肽的基因序列1)将连接后的多肽还原为核酸序列,并进行密码子优化;2)使用固相合成法合成抗原表位肽的基因序列;或由技术服务公司进行合成;4、慢病毒包装将上步合成的基因序列构建表达抗原表位肽的慢病毒表达质粒后,进行慢病毒包装;5、转染抗原递呈细胞(apc)并与pbmc共培养1)使用表达抗原表位肽的慢病毒转染抗原递呈细胞(包括但不限于:外周血单个核细胞、树突状细胞、中性粒细胞、b淋巴细胞、巨噬细胞);2)收集处理完成的apc,以apc:pbmc=1:5-20的比例混合共培养,得到效应细胞;6、筛选有效的精准多肽,并使用精准多肽再次刺激t细胞1)离心收集以上方案获得的t细胞,多肽作为抗原直接刺激t细胞筛选精准多肽:2)设置阳性对照:t细胞+100ng/mlokt3;阴性对照:t细胞+1640+10%fbs+200u/mlil2;3)精准多肽评判标准:阳性对照和阴性对照正常,则说明此数据可信;实验组显著大于阴性对照组的为有效的精准多肽;4)以筛选出的精准多肽二次冲击t细胞;7、构建tcr-t细胞1)对刺激后的t细胞进行cd8、cd137、ifn-γ的染色,并以流式细胞仪进行分选;2)分选出能够识别精准多肽的特异性细胞,测序确定高频tcr序列并扩增;3)构建tcr基因表达载体,包装病毒;4)敲除所述外周血t细胞中原有的tcr基因,转入上步构建的tcr基因,培养即得tcr-t细胞;8、免疫抑制性信号经抗体药物体外封闭,即得lrfft2细胞细胞表面抑制性信号分子包括:pd-1、tim-3、lag3、ctla-4、btla、vista、cd160、2b4(cd244);9、构建特异性抗原表达靶细胞及肿瘤模型生存实验。本发明的有益效果:1.肿瘤抗原为突变抗原,与其它组织不同,靶点专一性强,不易发生脱靶效应,安全性高;2.获得的特异性细胞比例高,通常能够识别肿瘤抗原的特异性细胞,在pbmc的分布为0.5%以下,经过lrfft2方案改造的细胞,识别肿瘤抗原的特异性t细胞(tcr+)比例为70%以上;3.lrfft2细胞由于对pd1、ctla4、tim3、lag3等免疫抑制性靶点进行敲除,因此,对肿瘤的杀伤能力不受限制,杀伤效率更高。附图说明图1:慢病毒转染apc效率检测;其中,1a:对照组,1b:转染组。图2:lrff细胞分型检测。图3:精准多肽的筛选。图4:流式检测特异性t细胞比例;其中,4a:对照组,4b:lrff方案。图5:tcr分布频率。图6:原有tcr的敲除效率检测;其中,6a:敲除前,6b:敲除后。图7:特异性tcr的表达效率;其中,7a:转染前,7b:转染7天后。图8:免疫抑制性信号的体外封闭效果;其中,8a:封闭前,8b:封闭后。图9:ldh释放检测杀伤效率。图10:elisa检测细胞因子ifn-γ的释放。图11动物荷瘤模型生存曲线。具体实施方式下面通过具体的实施方案叙述本发明。除非特别说明,本发明中所用的技术手段均为本领域技术人员所公知的方法。另外,实施方案应理解为说明性的,而非限制本发明的范围,本发明的实质和范围仅由权利要求书所限定。对于本领域技术人员而言,在不背离本发明实质和范围的前提下,对这些实施方案中的物料成分和用量进行的各种改变或改动也属于本发明的保护范围。技术方案详述如下:1、抗原表位预测1)使用肺癌患者外周血进行ctdna测序和hla分型检测;2)利用软件对测序信息进行分析:将ctdna测序结果与正常细胞的基因组相比,筛选出突变位点;3)以突变的氨基酸位点为中心,向两侧延伸8个氨基酸,将这段17个氨基酸的多肽作为潜在抗原表位;4)使用预测软件分析潜在抗原表位的ic50(推荐软件:netmhcpan3.0、pickpocket、artificialneuralnetworks(ann)),如ic50<1000nm则认为此潜在抗原表位为抗原表位。2、多肽连接1)使用前述软件分析任意抗原表位两两相连后接头处的ic50,ic50≥1000nm时认为是弱免疫原性,可以连接;ic50<1000nm时认为是强免疫原性,不能连接(此处应考虑3个预测软件的ic50计算结果,当≥2个软件计算的ic50≥1000nm时才能认为是弱免疫原性,当≥2个软件计算的ic50<1000nm时才能认为是强免疫原性);2)根据上述结果,将抗原表位连接在一起,接头处ic50要高于两侧抗原表位的ic50(也就是接头处尽量避免产生强结合抗原);必要时将弱免疫原性肽做为连接肽将强免疫原性肽间隔;或者添加患者自身氨基酸到接头处,用来降低产生强抗原的可能性。3、合成编码多肽的基因序列1)将连接后的多肽还原为核酸序列,并进行密码子优化;连接完成后的核酸序列如果较短(<100bp)可以适当把氨基酸序列进行重复,但是,需要注意还原成基因序列时,应尽量避免基因序列中反向重复、直接重复和镜像重复序列的出现2)使用固相合成法合成抗原表位肽的基因序列(由技术服务公司进行合成)。4、慢病毒包装1)将合成的抗原表位肽基因序列克隆到pcdh-mscv-mcs-ef1-copgfp质粒中,构建表达抗原表位肽的慢病毒表达质粒;2)慢病毒包装:a.复苏293t细胞,传两代后用于慢病毒包装;b.细胞转染:(t175培养瓶)取一支15ml离心管(标记为a),将400μllipofectamine2000轻轻地加入4mldmem中,轻轻地混匀,室温静置培养5min;另取两支15ml离心管(标记为b和c,b为对照组,c为实验组),加入以下试剂并轻轻地混匀,室温静置培养5min;将a管液体平均转入到b管和c管内,轻轻地混匀,室温静置培养20min。将t175中旧的培养基倒出,使用pbs把细胞洗一遍,换成新的25mldmem(不含抗生素和血清),轻轻地加入a、b或a、c混合液,轻轻摇匀,置于37℃,5%co2培养箱中培养;转染6h后,吸去含有转染复合物的培养基,更换为37℃预热的新鲜培养基;培养48h,并收集;3)慢病毒浓缩及滴度测定:慢病毒包装成功后,收集慢病毒上清液,4℃,4000g离心10min;用0.45μm滤器过滤上清液,除去细胞碎片;按照病毒上清液:浓缩试剂=5:1的比例混合,4℃放置2h或者过夜;将孵育好的混合液于4℃,4000g离心30min,即可见管底有米白色沉淀;小心移去上清(切勿碰触沉淀物);加入适量体积的dmem或者pbs,轻轻吹打重悬沉淀物;按需求分装病毒,-80℃保存(注:慢病毒切忌反复冻融,每冻融一次,慢病毒效价将下降10%-20%);测定前一天,将生长状态良好的293h细胞消化计数后稀释至5×104/ml,加入96孔板,100μl/孔,为每个病毒准备8-10个孔。放入37℃,5%co2培养箱中培养取一定量的病毒液感染细胞:在ep管中做10倍梯度稀释。稀释方法如下:每种病毒准备10个1.5mlep管,每管加入90μl培养液,往第一个管中加入10μl病毒原液,记作100;混匀后,吸取10μl加入第二个管混匀,记作10-1;以此类推(100-10-8),在对应细胞孔中加入10μl稀释好的病毒液并做好标记,培养48-72h后观察结果;滴度计算:对于带有荧光标记的慢病毒可使用荧光技术法测定滴度;在荧光显微镜下观察结果,并数出最后两个有荧光的荧光细胞克隆数,假设为x和y,则滴度(tu/ml)=(x+y×10)×1000/2/x孔的病毒液的含量(μl)。5、慢病毒转染抗原递呈细胞(apc)并与pbmc共培养1)使用rpmi-1640配制含有300u/mlril-2,10%fbs的全细胞培养基,记为rpmi-10-il-2;使用rpmi-10-il-2把apc细胞浓度调整到1×106/ml;使用表达抗原表位肽的慢病毒以moi=5-20感染apc(包括但不限于:外周血单个核细胞、树突状细胞、中性粒细胞、b淋巴细胞、巨噬细胞);放入37℃培养72h;4)收集处理完成的apc,以apc:pbmc=1:5-20的比例混合,pbmc约为5×107,加入50mlokm100培养基到t75培养瓶中;放入30-37℃细胞培养箱中培养14天,即获得lff方案效应细胞。6、筛选有效的精准多肽多肽作为抗原直接刺激效应细胞筛选精准多肽:1)离心收集以上lff方案细胞,1500rpm离心5min收集t细胞,加入10mlpbs重悬细胞并计数,1500rpm离心5min,收集t细胞以1640+10%fbs+200u/mlil2重悬,计数调整至1×106cells/ml;2)以排枪将t细胞分至96孔平底板中,200μl/孔,细胞数为2×105cells;再分别加入10μl1mg/ml的突变多肽,终浓度为50μg/ml,每条多肽设置3复孔;3)设置阳性对照:t细胞+100ng/mlokt3;阴性对照:t细胞+1640+10%fbs+200u/mlil2;4)37℃、5%co2刺激24h后,1500rpm离心10min,转移140μl上清至新的96孔板中;5)再将96孔板进行离心,1500rpm10min,取样品进行elisa检测(或将样品至于-80℃保存);检测ifn-γ的elisa系统:1)目前测试过可用于检测ifn-γ的elisa试剂盒有biolegend:legendmaxhumanifn-γelisakitwithpre-coatedplates(货号:430107)和达科为:humanifn-γelisakit(货号:dkw12-1000-096),请严格按照厂家说明书进行操作;2)elisa手动包板系统(15块板):humanifn-gammaduoset15plate(货号:dy285b)×1,duosetelisaancillaryreagentkit2(货号:dy008)×3;精准多肽评判标准:1)阳性对照和阴性对照正常,则说明此数据可信;2)实验组显著大于阴性对照时,说明多肽为有效的精准多肽。7、以筛选出的精准多肽二次冲击t细胞1)pbmc以步骤5培养至第2~14天时,取2×107的效应细胞,加入终浓度为10μg/ml~100μg/ml的精准多肽,冲击1-4h;2)冲击4h后,转入okm25预包板的6孔板中或t25cm2培养瓶,补okm100+12%fbs,37℃5%co2培养,根据细胞生长情况,转移至t75培养瓶中,尽量保持细胞密度在1×106cells/ml;3)进入t175培养瓶中时,培养基为okm200+5%fbs,培养10~14天即可获得精准多肽二次冲击得到的t细胞,即lrff细胞;8、突变抗原特异性杀伤t细胞的培养及分离1)以筛选出的精准多肽直接作为抗原刺激,对步骤7获得的lrff细胞进行刺激,刺激12~72h后,备用;2)对刺激后的t细胞进行cd8、cd137、ifn-γ的染色,并以流式细胞仪进行分选,选择cd8+cd137+、或者cd8+ifn-γ+细胞;9、cd8+t细胞tcr频率检测及高频tcr的克隆1)提取分选的cd8+cd137+、或者cd8+ifn-γ+细胞的基因组,进行tcr频率的检测,确定高频的tcr序列;2)提取分选细胞的mrna,反转录成从dna,根据高频tcr的序列,设计引物,扩增得到tcr基因;3)构建tcr基因表达载体,包装病毒;10、构建tcr-t细胞1)复苏pbmc,以磁珠分选cd8+细胞,备用;2)以crispr技术对cd8+t细胞原有的tcr进行基因敲除,检测无tcr表达时,再转入构建的tcr表达载体;3)将转入tcr基因的cd8+t细胞进行扩增培养,培养至10~21天时,即收获tcr-t细胞。11、免疫抑制性信号的封闭1)免疫抑制性信号分子包括:pd-1、tim-3、lag3、ctla-4、btla、vista、cd160、2b4(cd244);2)1000rpm离心5min,收集培养好的tcr-t细胞;3)以pbs洗一次,1000rpm离心5min,以okm-200+5%fbs重悬tcr-t,并调整至1×107/ml;4)加入抑制性信号分子的单抗药(如pd1/pdl1),终浓度为50~200μg/ml,优选150μg/ml,0~37℃封闭1~4h,优选37℃1h,即得lrfft2细胞。12、构建特异性抗原表达靶细胞及肿瘤模型生存实验1)构建可以表达筛选的精准多肽(特异性抗原)的慢病毒载体。2)将特异性抗原表达慢病毒载体包装成慢病毒颗粒,感染hla配型合适的肿瘤细胞,稳定过表达特异性抗原,流式检测表达水平及表达强度。3)稳定过表达特异性抗原肽的肿瘤细胞系接种ngs小鼠,做异位荷瘤动物模型。将5×105表达特异性抗原的肿瘤细胞悬于100μl生理盐水中,分别皮下注射至30只nsg小鼠的右侧胁肋部皮下,同时对小鼠进行编号。4)在肿瘤生长至100-120mm3左右时分组回输细胞,根据肿瘤体积大小,将动物模型随机分成三组,每组5-6只小鼠,一组给予安慰剂生理盐水,一组给予没有进行任何遗传操作的t细胞(对照组)1×107,一组给予lrfft2细胞1×107,首次注射细胞7天后进行第二次注射,7天之后第三次注射细胞,连续观察60天,统计存活数据,绘制生存曲线。试验结果:1、突变位点及抗原表位预测表1为测序检测到的突变位点及抗原表位预测结果;表1抗原表位预测2、慢病毒转染apc效率检测1)使用rpmi-1640配制含有300u/mlril-2,10%fbs的全细胞培养基,记为rpmi-10-il-2;使用rpmi-10-il-2把apc细胞浓度调整到1×106/ml;使用表达抗原表位肽的慢病毒以moi=5-20感染apc(包括但不限于:外周血单个核细胞、树突状细胞、中性粒细胞、b淋巴细胞、巨噬细胞);放入37℃培养72h;2)使用流式细胞仪检测apc中gfp阳性比例(如图1所示)。3、lff细胞分型检测lff方案细胞培养结束后,进行cd4+和cd8+细胞的分型检测,结果如图2所示:cd8+t细胞为89.1%,cd4+t细胞为8.11%。4、以lff细胞筛选精准多肽以12条多肽分别刺激培养的t细胞,通过检测ifn-γ的分泌,检测有效的多肽,结果如图3所示:3号和7号多肽引起的ifn-γ的释放量>阴性对照的释放量,属于有效精准多肽。5、对精准多肽特异性的t细胞的鉴定及分选以筛选的3号和7号多肽,刺激lff方案细胞,以流式检测对精准多肽特异性的t细胞比例,结果如图4所示,fl1+为特异性t细胞:lrff方案细胞,3号和7号多肽引起的释放ifn-γ的细胞比例,明显高于没有刺激的细胞(对照),说明,lrff方案,可以获得对精准多肽的特异性t细胞;同时以流式细胞仪进行cd8+ifn-γ+细胞(fl1+)的分选;6、高频tcr的鉴定及克隆将分选得到细胞进行基因组的提取,及tcr的测序,tcr的分布情况如图5所示(高频分布的前20),tcr6分布频率较高,说明,此tcr与突变抗原密切相关,根据tcr序列,对tcr进行扩增,构建慢病毒表达载体。表2tcrβ链cdr3的序列情况已知的tcr-α:氨基酸序列:mmkslrvllvilwlqlswvwsqqkeveqnsgplsvpegaiaslnctysdrgsqsffwyrqysgkspelimfiysngdkedgrftaqlnkasqyvsllirdsqpsdsatylcavnfgggklifgqgtelsvkpn碱基序列:已知tcr-β:氨基酸:mrirllccvafsllwagpviagitqaptsqilaagrrmtlrctqdmrhnamywyrqdlglglrlihysntagttgkgevpdgysvsrantddfpltlasavpsqtsvyfcasslsfgteaffgqgtrltvv横线为cdr3序列,需要被替换的序列替换后的tcr-β:横线为替换的cdr3序列7、原有tcr敲除效率的检测利用crispr技术,将pbmc上的原有的tcr进行敲除,结果如图6所示:可有效的减少原有tcr的表达,此时,可进行表达特异性tcr慢病毒的转染。8、特异性tcr表达情况的检测以包装特异性tcr的的慢病毒转染pbmc,在第7天时,以流式检测tcr的表达效率,结果如图7所示:构建的tcr可以正常表达,tcr+的细胞比例为25.1%9、免疫抑制性信号封闭效果在pbs缓冲体系中加入150μg/ml的荧光标记的单抗药keytruda,结合情况如图8所示,90%的细胞可以有效封闭。10、lrfft2细胞对靶细胞的杀伤作用分别以对照细胞和lrfft2细胞对突变抗原表位来源的靶细胞进行杀伤效率的检测,以无处理的细胞作为对照(mock),效靶比设置为40:1,结果如图9所示,与对照组相比,lrfft2细胞对靶细胞有较强的杀伤效果。11、lrfft2细胞释放细胞因子的检测肿瘤细胞与效应细胞共培养时,由于效应细胞,可以识别肿瘤细胞上突变抗原,因此,会产生一系列的细胞因子,ifn-γ是抗肿瘤作用中最主要的细胞因子之一,图10为lrfft2细胞与肿瘤细胞1:1共培养时,释放的ifn-γ的检测,结果表明:与效应细胞自身产生的ifn-γ相比(tcellsonly),与肿瘤细胞共培养后,lrfft2细胞可产生大量的ifn-γ此结果与杀伤实验结果一致说明:表达特异性tcr的t细胞,结合抑制性靶点的敲除可以更有效的提高抗肿瘤能力。13、构建特异性抗原表达靶细胞及肿瘤模型生存实验成功构建了特异性抗原表达肿瘤靶细胞系,建立荷瘤动物模型,结果显示(图11),lrfft2细胞对肿瘤荷瘤小鼠的生存改善具有显著影响作用。14、临床案例:某男:55岁疾病诊断:肝内胆管癌术后复发;第一疗程:每个月一次lrfft2细胞,数量1×109个细胞,共2次;第二疗程:每半年一次lrfft2细胞,数量1×109个细胞,共2次;给药结束后,22个月无进展生存;其它案例:患者编号疾病诊断无进展生存时间1胃腺癌肝转移2017.3-至今2胃癌2017.3-至今3肺癌2017.4-至今4肺腺癌2017.4-至今5肺腺癌2017.6-至今6食管癌2017.6-至今注:“至今”的含意为“申请日前一天”。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1