一种炔、胺和醛多组分聚合制备聚炔丙基胺的方法与流程
本发明涉及高分子化学和材料学领域,具体涉及一种炔、胺和醛的多组分聚合制备聚炔丙基胺的方法。
背景技术:
炔丙基胺在有机合成反应中是重要的中间体和骨架,以它为底物能够构建五元、六元、七元、八元含氮结构,目前已成功合成吡咯、吡咯烷、中氮茚、异吲哚、咪唑啉二酮、恶唑烷酮等分子。此外,炔丙基胺类物质对单体胺氧化酶具有抑制作用,具有良好的有效性和很高的选择性。合成炔丙基胺的方法被广泛的研究,但这些方法都有一些明显的缺点,如需要使用金属试剂、对水分敏感、环境耐受性差、反应底物不普遍适用于挥发性单体等。因此,开发出一种更绿色、简单、经济的方法来合成炔丙基胺化合物是很有必要的。
本发明通过一锅法多组分反应(mcr)的方法制备聚炔丙基胺具有操作简便、转化率高、原子经济、副产物少、条件温和等优点,并且符合绿色化学反应的特点。
技术实现要素:
为了解决现有技术的缺点和不足之处,本发明的目的在于提供一种炔、胺和醛的多组分聚合制备聚炔丙基胺的方法。
本发明的目的通过以下技术方案实现:
一种炔、胺和醛的多组分聚合制备聚炔丙基胺的方法,包括以下步骤:
1)在惰性氛围下,在有机溶剂中将炔、胺、硼酸与甲醛或炔、胺与甲醛进行反应,后续处理,获得聚炔丙基胺。
当炔、胺、硼酸与甲醛进行反应时,炔为r1-c≡c-cooh,胺为端基为nh2的二元胺,所述胺为h2n-ch2-r2-ch2-nh2;硼酸为
或者炔为ch≡c-r4-c≡ch,胺为端基为nh2的二元胺,所述胺为h2n-r2-nh2;硼酸为
当炔、胺与甲醛反应时,炔为hooc-c≡c-r4-c≡c-cooh,胺r6-hn-r2-nh-r6;r1、r2、r3、r4、r5、r6为有机基团。
作为优选,r1、r5、r6选自以下化学结构式中19~26中的任意一种,r2、r3、r4选自以下化学结构式1~18中的任意一种;
其中,m、h、k为1~20的整数,x选自o或s元素;*表示取代位置。
当炔、胺、硼酸与甲醛进行反应,炔为r1-c≡c-cooh时,炔、胺、醛、硼酸的摩尔比为2:1:4:(1~4);
当炔、胺、硼酸与甲醛进行反应,炔为ch≡c-r4-c≡ch时,反应中需加入催化剂;炔、胺、醛、硼酸的摩尔比为1:1:4:(2~6);所述催化剂为醋酸亚铜、碘化铜、氯化铜、溴化铜、醋酸铜、氧化亚铜、氧化铜中一种以上;
当炔、胺与甲醛反应,炔为hooc-c≡c-r4-c≡c-cooh时,炔、胺、醛的摩尔比为1:1:(2~6)。
所述反应的温度为45~80℃;所述甲醛以甲醛水溶液的形式加入;甲醛水溶液的浓度为37wt%~40wt%。
所述有机溶剂为1,2-二氯乙烷(dce)、间苯二甲腈(dcb)、四氢呋喃(thf)、甲苯、乙腈和1,4-二氧六环中的一种以上。
所述反应的时间为8~18小时。
所述后续处理是指将反应后的溶液加入沉淀剂进行沉淀,将沉淀物干燥。所述沉淀剂为甲醛、乙醚、乙腈、正己烷中的一种。
所述反应后的溶液以滴加的方式加入沉淀剂中,反应后的溶液加入沉淀剂中之前先用四氢呋喃进行稀释。
本发明的制备方法的反应方程式如式(i)~(iii):
反应式(i)~(iii)中,n为2-200之间的整数,r1、r2、r3、r4、r5、r6如前面炔、胺、硼酸所定义。
本发明的制备方法及所得到的产物具有如下优点及有益效果:
(1)本发明的制备方法反应原料易得,可通过购买或简单的反应制备;聚合条件温和,操作简便、转化率高、原子经济、副产物少。
(2)本发明制备得到的聚炔丙基胺化合物具有较高的折光指数,高于一些商业化有机聚合物材料,同时具有很好的成膜性。
附图说明
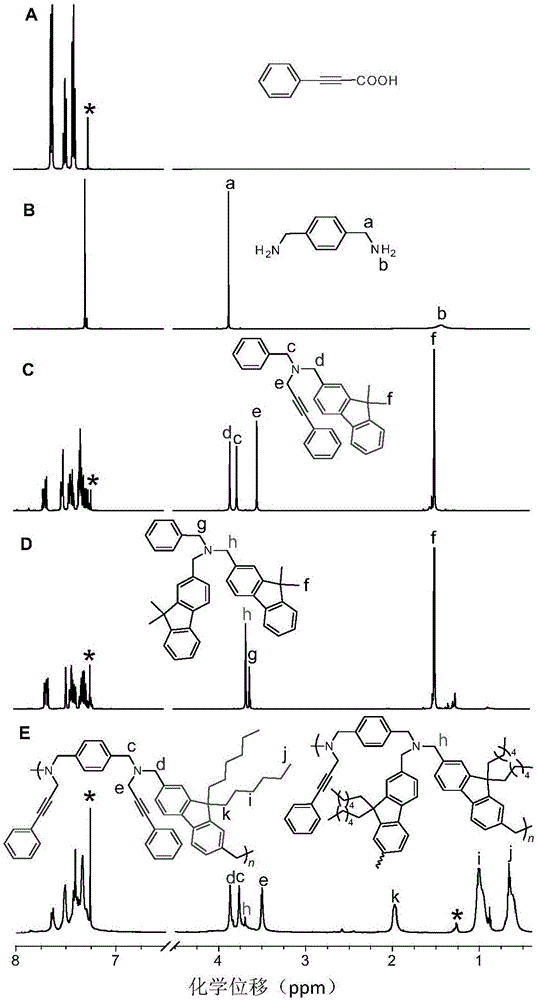
图1为实施例1制备的聚炔丙基胺化合物及其相应单体、模型化合物在cdcl3中核磁共振氢谱对比图;图c和d分别是两种模型化合物,e中左边化合物为聚炔丙基胺化合物,右边为产物中副产物;
图2为实施例1制备的聚炔丙基胺化合物及其相应单体、模型化合物的红外谱图;a为苯丙炔酸,b为1,4-苯二甲胺,c、d、e为对应的模型分子以及聚合物;
图3为实施例1制备的聚炔丙基胺化合物的热失重曲线图;
图4为实施例1制备的聚炔丙基胺化合物的折射率曲线图。
具体实施方式
下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
实施例1
本实施例的一种炔、胺和醛的多组分聚合制备聚炔丙基胺的方法,包括以下步骤:
(1)在10ml聚合管中加入0.07g(0.5mmol)1,4-苯二甲胺、0.15g(1.0mmol)苯丙炔酸和0.21g(0.5mmol)9,9-二己基芴-2,7-二硼酸,抽真空充氮气三次,接着向反应体系加入2.5mldce和0.2ml(2.0mmol)甲醛水溶液(甲醛的摩尔量为2mmol),加热至45℃反应10小时;
(2)反应完毕后冷却至室温,反应混合液用0.5mlthf稀释,再逐滴滴入(2滴/秒)装有50ml正己烷并以400转/分钟搅拌的锥形瓶中,得到黄色絮状沉淀,静置4小时后,过滤,得到的固体用20ml甲醇清洗3次,在40℃下真空干燥至恒重后得到0.30g淡黄色聚合物,产率为83%,分子量为13900g/mol。
本实施例中的所有反应单体均可通过购买得到。
本实施例制备得到的聚二炔丙基胺化合物具有p1结构式:
本实施例所涉及的反应方程式如下:
本实施例制备得到的聚炔丙基胺化合物在氘代氯仿中的氢谱核磁谱图如图1所示(a为苯丙炔酸,b为1,4-苯二甲胺,图c和d分别是两种模型化合物,e中左边化合物为聚炔丙基胺化合物,右边为产物中副产物)。从图1可看出,氘代氯仿的溶剂峰和水峰分别位于7.56ppm和1.56ppm,其中,h峰为杂质聚合物的亚甲基峰,c、d、e、均为制备得到的聚炔丙基胺化合物氢原子的出峰。
本实施例中制备得到的聚炔丙基胺化合物的红外吸收谱图如图2所示,a为苯丙炔酸,b为1,4-苯二甲胺,c、d、e为对应的模型分子以及聚合物(分别为图1中的c、d、e)。从图2可知,1,4-苯二甲胺中的胺基中氢原子出峰3287cm-1没有在聚合物中出现;在模型分子c以及聚合物的红外吸收谱图中都能看到2210cm-1处的c≡c伸缩振动峰,这说明了反应的顺利进行;聚合物中,3216cm-1处出现了羟基的伸缩振动峰,这可能是反应中副产物上的羟基特征峰。
本实施例中制备得到的聚炔丙基胺化合物在氮气氛围下的热重曲线图如图3所示。图3表明所得聚合物失重5%时的热分解温度为146℃。
本实施例中制备得到的聚炔丙基胺化合物薄膜随波长而变化的折射率曲线如图4所示。图4表明所得的聚炔丙基胺化合物具有较高的折光指数,高于一些折光指数在1.49~1.59的商业化有机聚合物材料。
实施例2
本实施例将实施例1中的1,4-苯二甲胺改为1,3-苯二甲胺,底物投料比以及浓度不变,反应产率为85%,分子量为9200g/mol。
本实施例所涉及的反应方程式如下:
实施例3
本实施例将实施例1中的溶剂dce改为dcb,底物投料比以及浓度不变,反应产率为82%,分子量为6700g/mol。
实施例4
本实施例将实施例1中的9,9-二己基芴-2,7-二硼酸改为四苯乙烯基对二硼酸,投料为0.5mmol,底物投料比以及浓度不变,反应产率为71%,分子量为10600g/mol。
本实施例所涉及的反应方程式如下:
实施例5
本实施例将实施例1中的苯丙炔酸改为对甲基苯丙炔酸,投料为0.16g(1.0mmol),底物投料比以及浓度不变,反应产率为88%,分子量为5100g/mol。
本实施例所涉及的反应方程式如下:
本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!