一种多靶点激酶抑制剂化合物的晶型A及制备方法和含有其的药物组合物与流程
本发明属于医药
技术领域:
,具体地说,涉及一种新型多靶点激酶抑制剂化合物的新晶型;本发明还涉及所述新晶型的制备方法以及含所述新晶型化合物的药物组合物。
背景技术:
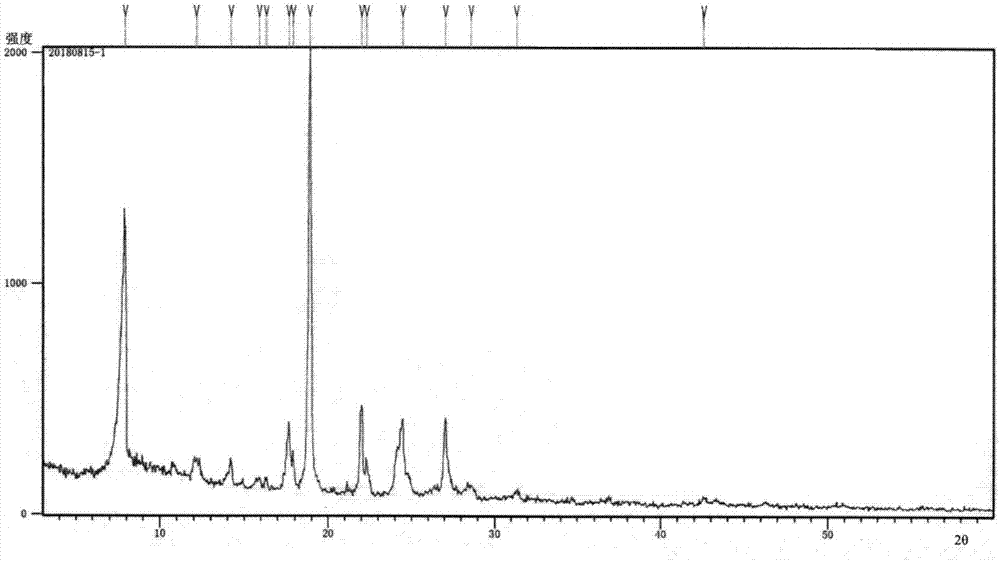
:蛋白激酶是一类催化蛋白质磷酸化反应的酶,而细胞内绝大多数重要的生命活动都与蛋白质的磷酸化有关,通过介导细胞信号转导过程,蛋白质的磷酸化调控了细胞命运,例如细胞的增殖、分化与凋亡。因此蛋白激酶已经成为热门的药物靶点,激酶抑制剂药物成为肿瘤靶向治疗最为重要的组成部分。激酶抑制剂作为药物目前已广泛应用于肿瘤靶向治疗、炎症治疗等领域,然而,随着激酶抑制剂的广泛使用,耐药问题已经成为当前临床面临的关键问题之一,研究资料显示,旁路代偿信号通路的激活是激酶抑制剂耐药的重要原因之一。发展可同时作用于多条信号通路的多靶点激酶抑制剂,不仅可有效应对肿瘤多分子异常的生物学特征,也能在一定程度上缓解药物耐药问题。对于一些异质性极大的疾病,多靶点药物可能是唯一有效的药物治疗方式,已被fda批准上市的sorafenib是一个多靶点激酶抑制剂,主要靶点包括c-raf、vegfr2、c-kit、p38α等,该药多靶点协同作用的机制可能是其对中晚期肝癌有效的重要原因。发展具有适度靶点选择性的多靶点激酶抑制剂具有一定的挑战性,以sorafenib为例,临床使用方面其有效性依旧不高,部分原因可能是其抑制了“抑制肝癌发展”的一些靶点,例如p38α。p38α被报道是一个肝癌抑制蛋白,因此,抑制它的活性明显不利于肝癌的治疗,且很可能带来意想不到的毒副作用。针对以上论述及存在的问题,本发明人设计发展了具有适度选择性的多靶点蛋白激酶抑制剂化合物(n-(3-氟-4-((2-(1-(2-羟基乙基)-1h-吡唑-4-基)噻吩并[3,2-b]吡啶-7-基)氧代)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺(中国专利cn108530464a化合物编号td32-4),从已有的实验数据来看,其在复杂异质性疾病的治疗和克服耐药方面具有非常大的前景。在此基础上,发明人进一步研究了该激酶抑制化合物的新晶型。在药学领域,已经有越来越多的研究将重点放在药物化合物的晶型上,同一药物的不同晶型在外观、溶解度、熔点、溶出度、生物有效性等方面可能会有显著不同,进而影响药物的稳定性、生物利用度及疗效。药物晶型对临床疗效的影响很大,主要是由于药物晶型对生物利用度的影响:例如,研究发现,那格列奈的s晶型与临床使用的h晶型溶解度均明显>b晶型;阿司匹林有晶型ⅰ和晶型ⅱ,相同给药剂量下服用ⅱ型的血药浓度超出ⅰ型达70%。通过掌握各种固体药物的晶型种类,可以保证药物晶型的稳定性,改善药物的溶出速率和生物利用度,从而提高临床疗效和安全性。上市药物sorafenib的现有制剂技术主要是通过制药工艺和辅料的选择与配比来提高溶出度(吴晓刚,药学与临床研究,p144-p147,apr;23(2)2015),本发明研究同类化合物的新晶型及其对药物稳定性、溶出度等的影响。技术实现要素:本发明的一个目的是提供一种化合物,即(n-(3-氟-4-((2-(1-(2-羟基乙基)-1h-吡唑-4-基)噻吩并[3,2-b]吡啶-7-基)氧代)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺(如式(1)所示)的a晶型;所述a晶型特征通过熔点测试、x-射线粉末衍射(xrd)、差示扫描量热分析(dsc)、热重分析(tg)、红外光谱(ir)进行了表征,该晶型具备制备药物制剂所需要的性能。本发明的另一个目的是提供该化合物新晶型的制备方法。本发明的再一个目的是提供含有所述晶型化合物的药物组合物。根据本发明的一个方面,先制备所述化合物(如式(1)所示)的粗品,然后通过重结晶的方法对该物质粗品进行结晶,获得化合物晶型。通过对该结晶进行熔点测量、x-射线粉末衍射、dsc、tg、ir等检测和分析,确证获得的结晶是一种新型的结晶,称为a晶型。具体地,当用cu辐射源进行x-射线粉末衍射时,所述的a晶型包括在位于2θ°为7.9±0.2°、18.9±0.2°的特征衍射峰,这些峰的相对强度(i/i0)均大于或等于30%,所述结晶在x-射线粉末衍射中还包含2θ°为17.6±0.2°、22.0±0.2°、24.4±0.2°、27.0±0.2°的特征衍射峰,这些峰的相对强度均大于或等于15%(见图1)。其中“±0.2°”为允许的测量误差范围。本发明的a晶型可以通过x-射线粉末衍射图谱进行表征。其特征在于其x射线粉末衍射图谱具有上述2θ°表示的特征衍射峰,如表1所示,特征衍射峰的相对强度接近下列数值。表1此处的术语“接近”是指相对强度测量值的不确定性。本领域技术人员理解相对强度的不确定性非常依赖于测量条件。相对强度值可以例如在±25%范围内改变或优选在±10%范围内改变。上述a晶型具有图1所示的x射线粉末衍射图谱。本发明采用差示扫描量热(dsc)技术对a晶型进行表征(见图2),其中差示扫描量热分析结果表明,供试样品在178.2℃开始溶解,检测温度范围180.5℃处存在吸热峰。本发明采用热重分析技术对a晶型进行表征(见图3),其中热重谱图(tg)显示在温度至194.0℃范围内失重量为1.23%;194.0℃至353.0℃范围内的失重量为33.18%;353.0℃至468.0℃范围内的失重量为13.87%,468.0℃至671.0℃范围内的失重量为51.71%。说明随着温度升高,该化合物发生降解。本发明的化合物的a晶型的红外图谱如图4所示,其中在3252、1678、1607、1549、1531、1500、1470、1429、1368、1285、1211、1053、829cm-1有较强吸收峰。根据本发明的另一方面,制备所述a晶型的方法包括:将(n-(3-氟-4-((2-(1-(2-羟基乙基)-1h-吡唑-4-基)噻吩并[3,2-b]吡啶-7-基)氧代)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺的粗品,加入到c1-c4烷基醇或者c3-c4烷基酮与(石油醚/正己烷/正庚烷)的混合溶剂中,加热回流至溶解;溶液澄清后开始降温,至析出固体,过滤收集固体,将收集的固体鼓风干燥即得a晶型。所述的醇选自甲醇、乙醇、丙醇、异丙醇和丁醇,优选乙醇;所述的酮选自丙酮、甲基乙基酮和正丁酮等,优选丙酮;酮与(石油醚/正己烷/正庚烷)的体积比(v/v)为1:1~10:1,优选2∶1;所述的粗品与溶剂的配比按g/ml计,重量体积比为1:5~40,优选为1∶25。将溶液优选加热到40~80℃,更优选c1-c4烷基醇加热至80℃;根据此实施方案,析固进行2~8小时,更优选为4小时。析固温度为0~40℃,优选10~20℃。析固完全后过滤,烘干温度为30~60℃,优选为50℃。根据本发明的又一方面,提供一种药物组合物,该药物组合物含有所述新晶型化合物和任选的药学上可接受的载体和/或赋形剂。所述药物组合物可进一步按照常规制剂方法配制成可供给药的形式,包括经口或胃肠外给药形式。在可供给药的形式中,应包含治疗有效量的a晶型。所谓“治疗有效量”是指在该剂量下,本发明的化合物能够改善或减轻疾病症状,或能够抑制或阻断疾病的发展。根据经验并考虑本领域的标准方法和参考文献,本领域技术人员可以很容易地选择各种载体和/或赋形剂并确定其用量。根据本发明的再一方面,本发明的晶型可单独用于制备治疗过渡性增殖疾病的药物,也可以和其他治疗药物联合制备,协同作用。本发明公开的(n-(3-氟-4-((2-(1-(2-羟基乙基)-1h-吡唑-4-基)噻吩并[3,2-b]吡啶-7-基)氧代)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺的a晶型,用于治疗过度增殖性疾病,所述的过度增殖性疾病主要指癌症,包括但不限于非小细胞肺癌、结肠直肠癌、顽固性非小细胞肺癌、胰腺癌、卵巢癌、乳腺癌、神经胶质瘤、脑癌或颈部癌症。本发明制备得到的(n-(3-氟-4-((2-(1-(2-羟基乙基)-1h-吡唑-4-基)噻吩并[3,2-b]吡啶-7-基)氧代)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺的新晶型,其具有稳定的形态,化学稳定性好,耐高温的特点,这种新晶型的(n-(3-氟-4-((2-(1-(2-羟基乙基)-1h-吡唑-4-基)噻吩并[3,2-b]吡啶-7-基)氧代)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺具备了制备制剂所需要的性能,且贮存方便,生产操作更为简便,质量更易控制,经试验验证,含有所述化合物晶型a的制剂配方组合物,15分钟溶出度均在80%以上。附图说明图1是本发明实施例1所得的新晶型a的x射线衍射图谱;图2是本发明实施例1所得的新晶型a的dsc图谱;图3是本发明实施例1所得的新晶型a的tg图谱;图4是本发明实施例1所得的新晶型a的ir图谱;图5是本发明实施例1所得的新晶型a的hplc图谱。具体实施方式所有原料和试剂均为商业购买。粗品制备:(n-(3-氟-4-((2-(1-(2-羟基乙基)-1h-吡唑-4-基)噻吩并[3,2-b]吡啶-7-基)氧代)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺粗品的制备,以碘化物ii、1-(4-氟苯基氨基甲酰基)-环丙烷羧酸为起始原料,参考专利申请(专利申请号cn201710121054.7)的方法来制备。【实施例1】取100g(n-(3-氟-4-((2-(1-(2-羟基乙基)-1h-吡唑-4-基)噻吩并[3,2-b]吡啶-7-基)氧代)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺粗品加入到反应瓶中,加入2500ml的乙醇,搅拌下升温回流至80℃。溶解后搅拌10min,再降温至20~25℃,至固体析出再搅拌析晶4h,抽滤。滤饼于50℃鼓风干燥,用五氧化二磷助干。得类白色固体89.6g,收率89.6%。用卡尔费休测定仪检测水分为0.1%。得到的化合物为(n-(3-氟-4-((2-(1-(2-羟基乙基)-1h-吡唑-4-基)噻吩并[3,2-b]吡啶-7-基)氧代)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺的a晶型。化合物性状鉴定如图1~图5所示。【实施例2】取100g(n-(3-氟-4-((2-(1-(2-羟基乙基)-1h-吡唑-4-基)噻吩并[3,2-b]吡啶-7-基)氧代)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺粗品加入到反应瓶中,加入2400ml的丙酮,搅拌下升温回流至60℃。溶解后搅拌10min,滴加石油醚(1200ml)至刚刚有固体析出,重新加热溶解。再降温至20~25℃,至固体析出再搅拌析晶4h,抽滤。滤饼于50℃鼓风干燥,用五氧化二磷助干,得到白色固体82.3g,收率82.3%。用卡尔费休测定仪检测水分为0.1%。经测试,所得到的化合物为(n-(3-氟-4-((2-(1-(2-羟基乙基)-1h-吡唑-4-基)噻吩并[3,2-b]吡啶-7-基)氧代)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺的a晶型。实施例样品的测试条件:(一)xrd:检测仪器:锐影(empyrean)x射线衍射仪检测条件:cu靶kα射线,电压40kv,电流40ma,发散狭缝1/32°,防散射狭缝1/16°,防散射狭缝7.5mm,2θ范围:3°-60°,步长0.02°,每步停留时间40s。检测依据:中华人民共和国(2015年版四部)0451x射线粉末衍射法。检测结果:如图1。(二)dsc:检测仪器:德国netzsch公司dsc214差示扫描量热仪检测条件:气氛:n2,40ml/min扫描程序:从室温以10℃/min升温至180℃,记录升温曲线。检测样品质量:样品1:2.26mg(使用铝质样品盘)检测依据:jy/t014-1996热分析方法通则检测结果:如图2。(三)tg:检测仪器:德国netzsch公司tg209f1热重分析仪检测条件:气氛:空气,20ml/min;扫描程序:室温~700℃,升温速率:10℃/min。检测依据:热分析方法通则jy/t014-1996检测结果:如图3。(四)红外光谱:检测仪器:ft-irirprestige-21(日本)检测条件:溴化钾压片法检测依据:gb/t6040-2002红外光谱分析方法通则检测结果:如图4。(五)hplc检测仪器:agilent1260infinityⅱ高效液相色谱仪(美国)检测条件:色谱柱:poroshell120ec-c18流动相a:水-流动相乙腈b(80:20)柱温:30℃检测波长:254nm。检测依据:《中国药典》二部附录vd高效液相色谱法检测结果:如图5。【实施例3】分别取20g(n-(3-氟-4-((2-(1-(2-羟基乙基)-1h-吡唑-4-基)噻吩并[3,2-b]吡啶-7-基)氧代)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺粗品加入到反应瓶中,参考实施例1的实验操作进行以下实验:表3实验结论:实验所用醇类溶剂,优选乙醇;实验所用酮类溶剂,优选丙酮;酮与(石油醚/正己烷/正庚烷)的体积比(v/v)为1:1~10:1,优选2:1;粗品与溶剂的配比按g/ml计,重量体积比为1:5~40,优选为1:25。将溶液优选加热到40~80℃,更优选c1-c4烷基醇加热至80℃;更优选烷基酮与(石油醚/正己烷/正庚烷)的混合溶剂加热至50℃;根据此实施方案,析固进行2~8小时,优选为4小时。析固温度为0~40℃,优选10~20℃。析固完全后过滤,烘干温度为30~60℃,优选为50℃。【实施例4】(n-(3-氟-4-((2-(1-(2-羟基乙基)-1h-吡唑-4-基)噻吩并[3,2-b]吡啶-7-基)氧代)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺的a晶型的稳定性考察将获得的a晶型进行稳定性考察(10天的加速试验),在60℃、湿度92.5%、光照条件下对新晶型的纯度、最大单杂及总杂与0天的数据进行对比,光照条件下,纯度略有降低,其他条件下显示获得的晶型稳定。表4a晶型影响因素试验结果将(n-(3-氟-4-((2-(1-(2-羟基乙基)-1h-吡唑-4-基)噻吩并[3,2-b]吡啶-7-基)氧代)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺的粗品进行稳定性考察(10天的加速试验),在40℃、60℃对粗品的纯度、最大单杂与0天的数据进行对比,粗品60℃5天、60℃10天的纯度低于a晶型相应条件下的纯度。表5粗品影响因素试验结果【实施例5】固体药物制剂的制备处方1:制法:上述成分按照常规制剂方法进行混合、直接压片。处方2:制法:将(n-(3-氟-4-((2-(1-(2-羟基乙基)-1h-吡唑-4-基)噻吩并[3,2-b]吡啶-7-基)氧代)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺a晶型化合物与甘露醇、乳糖、交联聚维酮按等量倍增法混合均匀,加入预先配好的hpmc溶液制成软材,20目筛制粒,60℃干燥30分钟,18目筛整粒,加入微粉硅胶,混合均匀,装入2#胶囊即可。处方3:制法:上述成分按照常规制剂方法进行混合、直接压片。【实施例6】影响因素对照试验按实施例5中的处方1~3工艺制备3批样品,经基本项目考察合格后,分别进行光照,高温和高湿试验,考察样品的外观性状、含量和溶出度。影响因素的结果表明,样品在高温和光照条件下性质稳定,可以作为制剂参考处方和工艺,但处方1~3在25℃、rh75%以及25℃、rh92.5%条件下稳定,在光照下不容易产生降解产物。其中处方3用粗品做成制剂,进行溶出度对比,结果如下:表4考察指标处方1处方2处方3溶出度好好差可压性好/差崩解度好较好差研制晶型的目的主要是解决溶出度的问题,增大溶出。按照2010版药典溶出度试验表明处方1~2其在15分钟溶出度均在80%以上。而处方3溶出度低于80%。在辅料一致的前提下,处方1的各项考察指标优于处方2,显著优于处方3。以上对本发明较佳实施方式的描述并不限制本发明,本领域技术人员可以根据本发明做出各种改变或变形,只要不脱离本发明的精神,均应属于本发明所附权利要求的范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1