一种香清兰苷中间体及其制备方法和应用与流程
本发明涉及医药研发领域,具体涉及一种香清兰苷中间体及其制备方法和应用。
背景技术:
香清兰苷(又称金合欢素-7-o-葡萄糖苷)有显著扩张冠状动脉的作用,可用于制备治疗心血管疾病,特别是抗心肌缺血的药物。香清兰苷对大鼠miri有明显的保护作用,有效减轻心肌病理学形态损伤,其机制可能与清除氧自由基有关,为临床治疗冠心病提供了可能。其结构如下式所示:
香清兰苷在植物中含量较高,但分离纯化较为繁琐。目前化学合成香清兰苷存在许多问题,例如以野漆树苷为原料,经氢化还原,相转移催化以及一水合胺脱保护四步反应得到香清兰苷(liu,jidanetal.carbohydrateresearch,357,41-46;2012),该路线收率较低,反应中溴代葡萄糖易产生副产物。
另外有报道运用碳酸银合成o-苷的方法,但该方法试剂价格高,反应条件严苛(tianyaoshietal.syntheticcommunications,41:17,2594-2600)。
因此探索一种能得到高纯度香清兰苷的化学合成的方法有重要意义。
技术实现要素:
本发明所要解决的技术问题在于为了克服现有香清兰苷的化学合成方法或存在收率低以及副产物的问题,或存在试剂成本高、反应条件严苛的问题,因而提供了一种香清兰苷中间体及其制备方法和应用。本发明的香清兰苷中间体可经一步脱保护反应直接转化为香清兰苷,并且其可由溴代苯甲酰基葡萄糖与5-o-己酰基金合欢素在较为温和方便和简洁的条件下得到,易于制备,避免了使用银盐作为催化剂;且反应收率高、无需柱层析纯化、对环境友好、试剂便宜、反应方便。
本发明是通过下述技术方案来解决上述技术问题的。
本发明提供了一种如式(i)所示的化合物,其结构如下:
其中,r为己酰基(hexanoyl)。
本发明进一步还提供了一种如式(i)所示的化合物的制备方法,其包括如下步骤:在碱和催化剂存在的条件下,将如式(iii)所示的化合物和如式(iv)所示的化合物进行取代反应,即可;
本发明中,所述的取代反应可采用本领域中此类取代反应的常规反应条件和参数;其中,r为己酰基。
本发明中,所述的取代反应的反应溶剂为本领域常规所用,本发明中具体可使用氯仿、水、n,n-二甲基甲酰胺、甲苯和乙醇中任意两种所形成的两相溶剂,例如氯仿:水=1:1-2:1(v/v)、dmf:水=1:1(v/v)、甲苯:乙醇=1:1(v/v)。
本发明中,所述的反应溶剂的用量为本领域常规所用,本发明中所述的如式(iii)所示的化合物与所述的反应溶剂的质量体积比可为1:3-1:5。
本发明中,所述的取代反应的反应温度为本领域常规所用,本发明中具体可使用30℃-50℃,例如40℃。
本发明中,所述的催化剂可为本领域常规所用的相转移催化剂,本发明中具体可使用四丁基溴化铵(tbab)、苄基三乙基氯化铵(teba)、四丁基氯化铵、和三辛基甲基氯化铵中的一种或多种。
本发明中,所述的催化剂的用量为本领域常规所用,本发明中所述的如式(iii)所示的化合物与所述的催化剂的摩尔比可为1:1-3:1。
本发明中,所述的碱为本领域常规所用,本发明中具体可使用碳酸钾、氢氧化钠和氢氧化钾中的一种或多种,更具体可为碳酸钾。
本发明中,所述的碱的用量为本领域常规所用,本发明中所述的如式(iii)所示的化合物与所述的碱的摩尔比可为1:1.5-1:2.5,或1:1.8-1:2.2,例如1:2。
本发明中,所述的取代反应的反应进程可采用本领域常规检测方式进行监测,如薄层色谱(tlc)、气相色谱(gc)、核磁共振波谱(nmr)或高效液相色谱(hplc)等;本发明优选采用tlc或hplc,当采用hplc作为监测手段时,优选以反应体系中所述的如式(iii)所示的化合物不再参与反应或其浓度低于0.5%作为反应终点。
本发明中,所述的取代反应的反应时间以上述监测过程中监测到所述的取代反应到达反应终点为准,本发明中具体可为20h-24h。
本发明中,所述的取代反应的反应物料的加料顺序可为本领域中此类反应常规所用。本发明优选将所述的反应溶剂、所述的如式(iii)所示的化合物、所述的如式(iv)所示的化合物依次加入反应体系中,再加入所述的碱和所述的催化剂,优选将所述的碱和所述的催化剂以滴加方式加入。
本发明中,所述的取代反应在反应到达终点后,优选所述的制备方法还包括下述后处理过程:将反应液中用稀盐酸(例如1mhcl水溶液)淬灭,并用ch2cl2与饱和食盐水洗涤,分离有机相并浓缩,即可。进一步优选在所述的后处理过程后还包括下述纯化过程:将浓缩物进行柱层析纯化,所用的洗脱液配比优选为pe:ea:ch2cl2=5:1:2。
本发明中,所述的如式(iii)所示的化合物为溴代苯甲酰基葡萄糖,其可采用本领域常规合成手段直接制备得到。
本发明中,所述的如式(iv)所示的化合物为5-o-己酰基金合欢素,其亦可采用本领域常规合成手段直接制备得到,本发明中具体可参考qigaoet.al.carbohydrateresearch,2009,344,511-515中报道的合成路线:将如式(v)所示的化合物先进行5位和7位己酰基保护得到如式(vi)所示的化合物,再脱除后者7位的己酰基保护基,即可;
其中,r为己酰基。
本发明进一步还提供了一种如式(iv)所示的化合物:
其中,r为己酰基。
本发明进一步还提供了一种如式(ii)所示的香清兰苷的制备方法,其包括如下步骤:将如式(i)所示的化合物进行如下式所示的脱保护反应,即可;
其中,r为己酰基。
本发明中,所述的脱保护反应可采用本领域中常规脱除葡萄糖环上氧原子的酰基保护基的常规反应条件和参数。本发明中并不作特别限定,例如可参考文献qigaoet.al.carbohydrateresearch,2009,344,511-515。
本发明中,所述的脱保护反应的反应溶剂为本领域常规所用,本发明中具体可使用甲醇、二氯甲烷、n,n-二甲基甲酰胺、四氢呋喃和丙酮中的一种或多种;例如可使用甲醇:二氯甲烷=1:1(v/v)。
本发明中,所述的反应溶剂的用量为本领域常规所用,本发明中所述的如式(i)所示的化合物与所述的反应溶剂的质量体积比可为1:30-1:40(g/ml)。
本发明中,所述的脱保护反应的反应温度为本领域常规所用,本发明中具体可使用20℃-30℃。
本发明中,所述的脱保护反应的反应试剂为本领域常规所用,本发明中具体可使用甲醇钠、氢氧化钠、和碳酸钾中的一种或多种,例如甲醇钠。
本发明中,所述的反应试剂的用量为本领域常规所用,本发明中所述的如式(i)所示的化合物与所述的反应试剂的摩尔比可为1:0.8-1:1.2。
本发明中,所述的取代反应的反应进程可采用本领域常规检测方式进行监测,如薄层色谱(tlc)、气相色谱(gc)、核磁共振波谱(nmr)或高效液相色谱(hplc)等;本发明优选采用tlc或hplc,当采用hplc作为监测手段时,优选以反应体系中所述的如式(i)所示的化合物不再参与反应或其浓度低于0.5%作为反应终点。
本发明中,所述的脱保护反应的反应时间以上述监测过程中监测到所述的脱保护反应到达反应终点为准,本发明中具体可为4h-8h。
本发明中,所述的脱保护反应的反应物料的加料顺序可为本领域中此类反应常规所用。本发明优选将所述的反应溶剂、所述的如式(i)所示的化合物、所述的反应试剂依次加入反应体系中,进一步优选在室温条件下依次加入。
本发明中,所述的脱保护反应在反应到达终点后,优选所述的制备方法还包括下述后处理过程:将反应液中析出的黄色固体抽滤,洗涤,干燥即可。其中所述的抽滤,洗涤,干燥均为本领域常规所述,所述的洗涤溶剂优选为甲醇,并优选洗涤两次。
进一步地,本发明的如式(ii)所示的香清兰苷的制备方法还包括如下步骤:将如式(iii)所示的化合物和如式(iv)所示的化合物进行取代反应,得到如式(i)所示的化合物;
其中,r为己酰基;所述的取代反应的反应条件和参数均如上所述。
本发明进一步还提供了一种如式(iii)所示的化合物、如式(iv)所示的化合物或如式(i)所示的化合物在制备香清兰苷中的应用。
在符合本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
本发明所用试剂和原料均市售可得。
本发明的积极进步效果在于:
1、本发明的香清兰苷中间体可经脱保护反应一步得到香清兰苷,在制备中间体的过程中避免了使用银盐作为催化剂、且反应收率高、无需柱层析纯化、对环境友好、试剂便宜、反应方便。
2、本发明的香清兰苷中间体可由溴代苯甲酰基葡萄糖与5-o-己酰基金合欢素在较为温和方便和简洁的条件下得到,易于制备。
附图说明
图1为由实施例1制备的如式(i)所示化合物的核磁氢谱。
图2为由实施例1制备的如式(i)所示化合物的核磁碳谱。
图3为由实施例7制备的如式(ii)所示化合物的核磁氢谱。
图4为由实施例7制备的如式(ii)所示化合物的核磁碳谱。
具体实施方式
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
制备例1如式(iii)所示化合物(溴代苯甲酰基葡萄糖)的制备
将28mmold-葡萄糖溶于30ml二氯甲烷,加入180ml吡啶,冰浴下滴加166ml苯甲酰氯,缓慢升温至室温后搅拌过夜,反应完全。反应液依次用1mhcl,饱和碳酸氢钠,饱和食盐水洗涤,干燥旋干后,用石油醚和乙酸乙酯重结晶得到白色固体,收率92%,纯度98.7%。
制备例2如式(iv)所示化合物(5-o-己酰基金合欢素)的制备
将0.02mmol金合欢素(式v化合物)溶于50mln,n-二甲基甲酰胺,加入6.9ml三乙胺,2mmol4-二甲氨基吡啶,冰浴下滴加0.05mmol十二酰氯,缓慢升温至室温后继续反应4h,加入100ml二氯甲烷,依次用1mhcl、饱和碳酸氢钠溶液、饱和食盐水洗涤,干燥旋干后,用甲醇结晶得到白色固体,该步收率87%,直接用于下步反应。
将5mmol5,7-二己酰基金合欢素(式vi化合物)溶于40ml甲醇-二氯甲烷(1:1)中,加入2.5mmol碳酸钾,室温下搅2.5h,冰浴下加入1mhcl/meoh溶液淬灭,旋干后经柱层析纯化(甲苯:乙酸乙酯=5:1)得到白色固体(iv),收率95%。
1hnmr(400mhz,cdcl3)δ7.70(d,j=8.9hz,2h,ar-h-2’,6’),6.90(d,j=8.9hz,2h,ar-h-3’,5’),6.76(d,j=2.2hz,1h,ar-h-3),6.55(d,j=2.3hz,1h,ar-h-6),6.50(s,1h,ar-h-8),3.84(s,3h),2.75(t,j=7.7hz,2h),1.87-1.76(m,2h),1.45-1.30(m,4h),0.90(t,j=7.1hz,3h);13cnmr(100mhz,cdcl3)δ177.51,173.32,162.54,162.43,161.78,158.74,150.49,127.85,123.16,114.37,110.21,109.36,105.98,101.37,55.44,34.32,31.33,24.14,22.36,13.91.hrms[m+na]+calcdforc22h22o6na405.1314,found405.1324.
实施例1如式(i)所示化合物(5-o-己酰基金合欢素-7-o-β-d-(2’,3’,4’,6’-四苯甲酰基)葡萄糖)的制备
在反应瓶中将0.8mmol5-o-己酰基金合欢素与1.2mmol2,3,4,6-四苯甲酰基溴代葡萄糖溶解在20ml氯仿中,另将2.4mmolk2co3,0.4mmoltbab溶于10mlh2o中,将该水溶液缓慢滴加在上述氯仿溶液中,40℃下搅拌24h反应完全,加入1mhcl淬灭反应,反应液用ch2cl2与饱和食盐水洗涤。有机相干燥旋干后得到油状物,柱层析(pe:ea:ch2cl2=5:1:2)得到如式(i)所示化合物(白色固体),收率86%,hplc纯度99.2%(hplc条件:waterssymmetryc184.6*150mm;流动相:0.01%三氟乙酸水溶液,0.01%三氟乙酸乙腈溶液,流速:0.8ml/min,检测波长275nm)。
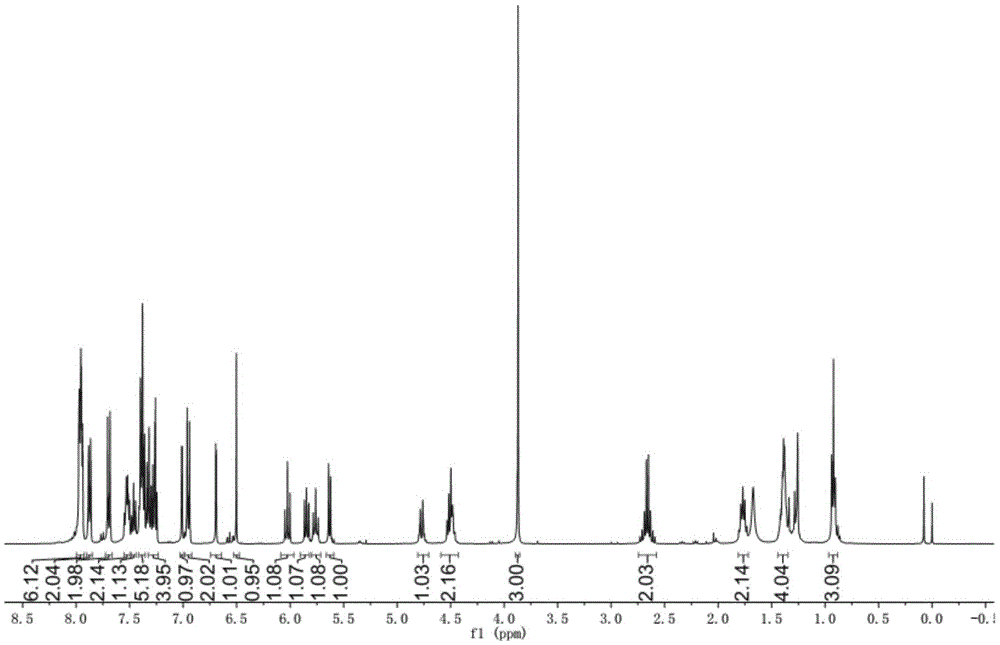
1hnmr(400mhz,cdcl3)δ7.87(d,j=8.7hz,2h,ar-h-2’,6’),7.25(d,j=8.7hz,2h,ar-h-3’,5’),7.17(d,j=2.1hz,1h,ar-h-8),6.78(d,j=2.1hz,1h,ar-h-6),6.59(s,1h,ar-h-3),5.79(d,j=2.4hz,0.8h,h-1α),5.54-5.44(m,1h,h-3),5.33(d,j=9.1hz,0.2h,h-1β),5.13(t,j=9.9hz,1h,h-4),4.35(dd,j=12.4,4.8hz,1h,h-5),4.07-3.97(m,2h,h-6),2.74(t,j=7.6hz,2h),2.59(t,j=7.5hz,2h),2.53(dd,j=13.2,5.3hz,1h,h-2),2.09(d,j=7.7hz,1h,h-2),2.08-2.02(m,9h),1.86-1.74(m,4h),1.49-1.34(m,8h),0.98-0.91(m,6h);13cnmr(100mhz,cdcl3)δ175.30,171.28,170.80,169.52,169.17,168.81,160.34,158.68,157.45,152.35,149.78,126.43,121.38,111.50,108.53,107.36,100.95,94.72,68.31,67.60,67.39,60.86,33.67,33.33,33.17,30.35,30.21,23.51,23.11,21.38,21.29,19.93,19.63,12.96,12.90.
实施例2如式(i)所示化合物(5-o-己酰基金合欢素-7-o-β-d-(2’,3’,4’,6’-四苯甲酰基)葡萄糖)的制备
在反应瓶中将0.8mmol5-o-己酰基金合欢素与1.2mmol2,3,4,6-四苯甲酰基溴代葡萄糖溶解在20ml氯仿中,另将1.6mmolnaoh,0.4mmoltbab溶于10mlh2o中,将该水溶液缓慢滴加在上述氯仿溶液中,40℃下搅拌24h反应,加入1mhcl淬灭反应,反应液用ch2cl2与饱和食盐水洗涤。有机相干燥旋干后得到油状物,柱层析(pe:ea:ch2cl2=5:1:2)得到如式(i)所示化合物(白色固体),收率为35.8%,纯度94.7%。
实施例3如式(i)所示化合物(5-o-己酰基金合欢素-7-o-β-d-(2’,3’,4’,6’-四苯甲酰基)葡萄糖)的制备
在反应瓶中将0.8mmol5-o-己酰基金合欢素与1.2mmol2,3,4,6-四苯甲酰基溴代葡萄糖溶解在20ml氯仿中,另将1.6mmolkoh,0.4mmoltbab溶于10mlh2o中,将该水溶液缓慢滴加在上述氯仿溶液中,40℃下搅拌24h反应,加入1mhcl淬灭反应,反应液用ch2cl2与饱和食盐水洗涤。有机相干燥旋干后得到油状物,柱层析(pe:ea:ch2cl2=5:1:2)得到如式(i)所示化合物(白色固体),收率45.7%,纯度为93.3%。
实施例4如式(i)所示化合物(5-o-己酰基金合欢素-7-o-β-d-(2’,3’,4’,6’-四苯甲酰基)葡萄糖)的制备
在反应瓶中将0.8mmol5-o-己酰基金合欢素与1.2mmol2,3,4,6-四苯甲酰基溴代葡萄糖溶解在20ml氯仿中,另将1.6mmolk2co3,0.4mmoltbab溶于10mlh2o中,将该水溶液缓慢滴加在上述氯仿溶液中,40℃下搅拌24h反应,加入1mhcl淬灭反应,反应液用ch2cl2与饱和食盐水洗涤。有机相干燥旋干后得到油状物,柱层析(pe:ea:ch2cl2=5:1:2)得到如式(i)所示化合物(白色固体),收率58.9%,纯度为95.3%。
实施例5如式(i)所示化合物(5-o-己酰基金合欢素-7-o-β-d-(2’,3’,4’,6’-四苯甲酰基)葡萄糖)的制备
在反应瓶中将0.8mmol5-o-己酰基金合欢素与1.2mmol2,3,4,6-四苯甲酰基溴代葡萄糖溶解在20ml氯仿中,另将3.2mmolk2co3,0.4mmoltbab溶于10mlh2o中,将该水溶液缓慢滴加在上述氯仿溶液中,40℃下搅拌24h反应,加入1mhcl淬灭反应,反应液用ch2cl2与饱和食盐水洗涤。有机相干燥旋干后得到油状物,柱层析(pe:ea:ch2cl2=5:1:2)得到如式(i)所示化合物(白色固体),收率49.7%,纯度为96.5%。
实施例6如式(i)所示化合物(5-o-己酰基金合欢素-7-o-β-d-(2’,3’,4’,6’-四苯甲酰基)葡萄糖)的制备
在反应瓶中将0.8mmol5-o-己酰基金合欢素与1.2mmol2,3,4,6-四苯甲酰基溴代葡萄糖溶解在20ml氯仿中,另将2.4mmolk2co3,0.4mmolaliquat336溶于10mlh2o中,将该水溶液缓慢滴加在上述氯仿溶液中,40℃下搅拌24h反应,加入1mhcl淬灭反应,反应液用ch2cl2与饱和食盐水洗涤。有机相干燥旋干后得到油状物,柱层析(pe:ea:ch2cl2=5:1:2)得到如式(i)所示化合物(白色固体),收率78%,纯度为97.6%。
实施例7如式(ii)所示化合物(香清兰苷)的制备
在反应瓶中将0.2mmol如式(i)所示化合物溶解于10mlch3oh中,加入0.12mmolch3ona。室温下反应过夜,析出黄色固体,抽滤,固体用甲醇洗涤两次,干燥得到香清兰苷,收率为90%,hplc纯度为99.6%(hplc条件:waterssymmetryc184.6*150mm;流动相:0.01%三氟乙酸水溶液,0.01%三氟乙酸乙腈溶液,流速:0.8ml/min,检测波长275nm)。
1hnmr(400mhz,dmso)δ7.95(d,j=8.8hz,2h),6.97(d,j=8.8hz,2h),6.86(s,1h),6.84(d,j=1.4hz,1h),6.44(d,j=1.8hz,1h),5.07(d,j=7.5hz,1h),3.71(d,j=10.7hz,1h),3.47-3.21(m,5h);13cnmr(100mhz,dmso)δ182.45,164.81,163.41,162.29,161.58,157.41,129.05,121.15,116.58,105.79,103.42,100.16,95.29,77.63,76.91,73.58,69.99,61.02.
实施例8如式(ii)所示化合物(香清兰苷)的制备
在反应瓶中将0.2mmol如式(i)所示化合物溶解于10mlch3oh中,加入0.12mmolkoh。室温下反应过夜,析出少量黄色固体,抽滤,固体用甲醇洗涤两次,干燥得到香清兰苷,收率为45.5%,纯度为93.2%。
实施例9如式(ii)所示化合物(香清兰苷)的制备
在反应瓶中将0.2mmol如式(i)所示化合物溶解于10mlch3oh中,加入0.12mmollioh。室温下反应过夜,析出少量黄色固体,抽滤,固体用甲醇洗涤两次,干燥得到香清兰苷,收率为65.5%,纯度为95.3%。
对比例1
将0.02mmol金合欢素(化合物1)溶于50mln,n-二甲基甲酰胺,加入6.9ml三乙胺,2mmol4-二甲氨基吡啶,冰浴下滴加0.05mmol乙酰氯,缓慢升温至室温后继续反应4h,加入100ml二氯甲烷,依次用1mhcl、饱和碳酸氢钠溶液、饱和食盐水洗涤,干燥旋干后,用甲醇结晶得到白色固体2,该步收率82.5%,直接用于下步反应。
将5mmol5,7-二乙酰基金合欢素(化合物2)溶于40ml甲醇-二氯甲烷(1:1)中,加入2.5mmol碳酸钾,室温下搅拌2.5h,冰浴下加入1mhcl/meoh溶液淬灭,旋干后经柱层析纯化(石油醚:乙酸乙酯=8:1)得到淡黄色固体3,收率38.6%。
在反应瓶中将0.8mmol5-o-乙酰基金合欢素(化合物3)与1.2mmol2,3,4,6-四苯甲酰基溴代葡萄糖溶解在20ml氯仿中,另将2.4mmolk2co3,0.4mmoltbab溶于10mlh2o中,将该水溶液缓慢滴加在上述氯仿溶液中,40℃下搅拌24h反应完全,加入1mhcl淬灭反应,反应液用ch2cl2与饱和食盐水洗涤。有机相干燥旋干后得到油状物,柱层析(pe:ea=6:1)得到白色固体4,收率68.8%,纯度98.7%。
在反应瓶中将0.2mmol化合物4溶解于10mlch3oh中,加入0.12mmolch3ona。室温下反应过夜,析出黄色固体,抽滤,固体用甲醇洗涤两次,干燥得到香清兰苷,收率为86%,纯度为99.6%。
在上述由化合物2制备化合物3的实验过程中,发明人分别在该反应进行30min、1h和2.5h时,将反应液取样0.5ml,加4ml甲醇稀释过滤后在同上的hplc条件下进样hplc检测,检测条件为:waterssymmetryc184.6*150mm;流动相:0.01%三氟乙酸水溶液,0.01%三氟乙酸乙腈溶液,流速:0.8ml/min,检测波长275nm)。
发明人在下表中提供了制备例2与对比例1的相关数据对比,从表1中可以看出:当采用乙酰基保护策略时,在脱保护反应一步中,随着反应进行会产生大量5、7位同时脱除乙酰基保护基的副产物。
表1脱保护反应一步的反应情况对比
其中,乙酰基保护策略下的反应主产物为
- 还没有人留言评论。精彩留言会获得点赞!