一种丙烯酸酯类聚合物包覆的碳纳米管及其制备方法与流程
本发明涉及先进纳米材料与纳米改性技术领域,具体涉及一种丙烯酸酯类聚合物包覆的碳纳米管及其制备方法。
背景技术:
自1991年日本科学家iijima发现碳纳米管(cnt)以来,碳纳米管由于其优异的力学、热学、电学等性能以及独特的结构,使其在复合材料领域具有巨大的应用价值。碳纳米管填充的复合材料在力学性能、导电性以及电磁屏蔽性等方面有明显的提升。然而,由于碳纳米管与高分子材料的不相容性以及其大的长径比和比表面积,碳纳米管与基体之间的界面粘结力差并且极易团聚,这影响了碳纳米管对复合材料的增强和改性效果。
对碳纳米管进行表面有机改性和聚合物包覆可以提高其在基体中的分散性和增加与基体之间的界面粘结力。dangetal.使用混酸对碳纳米管进行羧基功能化,随后利用羧基与含氟改性剂之间的化学反应使碳纳米管表面接上含氟基团,碳纳米管表面上的含氟基团与基体聚偏氟乙烯(pvdf)有相似的表面能从而提高了其在聚偏氟乙烯基体中的分散性(advmater2007,19,852–857)。但经强酸处理后的碳纳米管的结构受到破坏,因此这种处理方法降低了碳纳米管的力学和导电性能。chenetal.同样采用混酸对原始碳纳米管进行表面处理,随后依次在羧基化后的碳管表面接上氯化亚砜、乙二醇以及原子转移自由基聚合(atrp)引发剂2-溴异丁酰溴,最后实现甲基丙烯酸甲酯的原位自由基聚合制备出聚甲基丙烯酸甲酯包覆的碳纳米管(carbon2015,95,895-903)。这一方法不但破坏了碳纳米管的化学结构,并且所需处理过程多,效率较低。中国专利102181116a公开了一种碳纳米管与离子液体进行研磨混合后与聚丙烯酸酯类聚合物进行熔融混炼制备复合材料的方法。但这一方法通过离子液体与碳纳米管之间的共轭作用很难实现碳纳米管在聚合物中的完全分散。
大量的研究发现,多巴胺(da)在弱碱性的水溶液中可以发生自聚合,所产生的聚多巴胺(pda)几乎可以粘附于任何固体表面,这一反应条件温和并且粘附能力强,被广泛的应用于各类材料。聚多巴胺包覆的碳纳米管在水和大多有机溶剂中具有良好的分散性,而且聚多巴胺层表面含有许多活性官能团,如氨基和羟基,这为接枝上其他有机分子或聚合物提供了反应位点。
技术实现要素:
本发明的目的是为了解决现有的对碳纳米管改性过程中破坏碳纳米管的化学结构,或者聚合物包覆后的碳纳米管在复合材料中的分散性较差的问题,而提供一种丙烯酸酯类聚合物包覆的碳纳米管及其制备方法。
本发明首先提供一种丙烯酸酯类聚合物包覆的碳纳米管的制备方法,该方法包括:
步骤一:将碳纳米管和多巴胺加入到tris-hcl水溶液中搅拌,得到聚多巴胺包覆的碳纳米管;
步骤二:将步骤一得到的聚多巴胺包覆的碳纳米管分散在溶剂中,然后在冰浴下加入到反应容器中,在反应容器中加入三乙胺搅拌,然后再滴入含有2-溴异丁酰溴的二氯甲烷溶液搅拌,将温度升至室温继续反应,得到溴化碳纳米管;
步骤三:将步骤二得到的溴化碳纳米管作为引发剂、催化剂、配体和丙烯酸酯类单体加入到反应溶剂中,得到的混合物在冰浴下超声分散均匀后,对反应瓶抽真空-通氮气,完全除去瓶中氧气,随后在50-80℃下进行atrp聚合,得到丙烯酸酯类聚合物包覆的碳纳米管。
优选的是,所述的碳纳米管和多巴胺的质量比为1:(1-10);
优选的是,所述的聚多巴胺包覆的碳纳米管和含有2-溴异丁酰溴的二氯甲烷溶液中的2-溴异丁酰溴的质量比为1:(5-10)。
优选的是,所述步骤三中引发剂、催化剂、配体和丙烯酸酯类单体的质量比为1:1:1.2:(20-40)。
优选的是,所述催化剂为氯化亚铜或溴化亚铜。
优选的是,所述配体为2-联吡啶、n,n,n',n,'n”-五甲基二亚乙基三胺或四甲基乙二胺。
优选的是,所述丙烯酸酯类单体为甲基丙烯酸甲酯、甲基丙烯酸乙酯、甲基丙烯酸丁酯、甲基丙烯酸缩水甘油酯、丙烯酸甲酯、丙烯酸乙酯或丙烯酸丁酯。
优选的是,所述步骤三的反应溶剂为n,n-二甲基甲酰胺。
优选的是,所述的步骤三的反应时间为6-24小时。
本发明还提供上述制备方法得到的丙烯酸酯类聚合物包覆的碳纳米管。
本发明的有益效果
本发明提供一种丙烯酸酯类聚合物包覆的碳纳米管及其制备方法,本发明利用多巴胺在碱性水溶液中的自聚合性而粘附到碳纳米管表面,粘合力强,整个过程不需要对碳纳米管进行酸化等处理,保留了碳纳米管独特的结构和优异的性能,并且不使用溶剂,是一种环境友好型的碳纳米管表面改性方法。其次,本发明中聚多巴胺起到了桥梁的作用,灵活的运用了聚多巴胺层上存在的大量的氨基和羟基,为接上原子转移自由基聚合引发剂2-溴异丁酰溴提供了反应位点,进而引发丙烯酸酯类单体进行原位自由基聚合,实现对碳纳米管的有效包覆。本发明聚合物包覆后的碳纳米管可以实现填料在聚合物树脂基体中的均匀分散,改善填料与基体之间的界面结合性能,从而对高聚物的增强和制备出有优异性能的复合材料具有重要意义。
附图说明
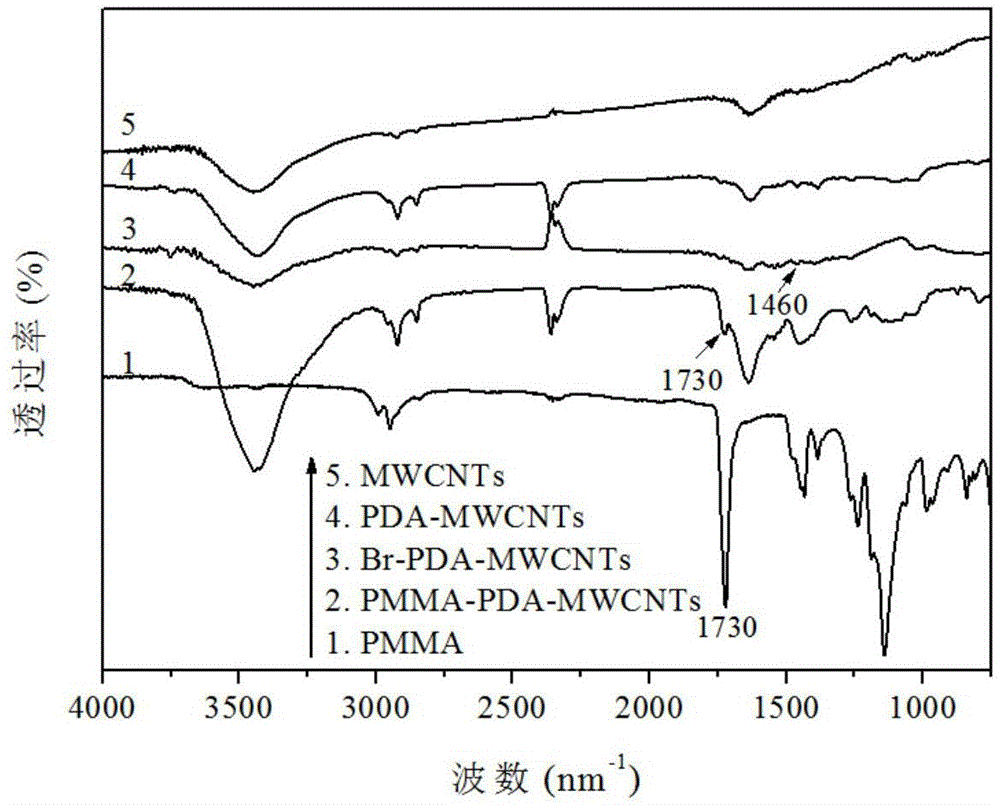
图1为原始碳纳米管、聚多巴胺包覆的碳纳米管、溴化碳纳米管、实施例1中聚甲基丙烯酸甲酯包覆碳纳米管以及聚甲基丙烯酸甲酯的傅里叶红外线光谱对比图。
图2为原始碳纳米管、聚多巴胺包覆的碳纳米管、溴化碳纳米管、实施例1中聚甲基丙烯酸甲酯包覆碳纳米管以及聚甲基丙烯酸甲酯的热失重曲线对比图。
图3为原始碳纳米管,聚多巴胺改性碳纳米管以及实施例1中聚甲基丙烯酸甲酯包覆碳纳米管的透射电镜图片。
图4为1wt%纯碳纳米管、实施例1中聚甲基丙烯酸甲酯包覆碳纳米管分散在聚偏氟乙烯基体中断面扫描电镜图片。
图5为纯聚偏氟乙烯以及含量为1wt%的纯碳纳米管、实施例1中聚甲基丙烯酸甲酯包覆碳纳米管填充聚偏氟乙烯复合材料的热失重曲线。
图6为原始碳纳米管、聚多巴胺包覆的碳纳米管、溴化碳纳米管、对比例1中聚甲基丙烯酸甲酯包覆碳纳米管以及聚甲基丙烯酸甲酯的热失重曲线对比图。
图7为原始碳纳米管,聚多巴胺改性碳纳米管以及对比例1中聚甲基丙烯酸甲酯包覆碳纳米管的透射电镜图片。
图8为1wt%纯碳纳米管、对比例1中聚甲基丙烯酸甲酯包覆碳纳米管分散在聚偏氟乙烯基体中断面扫描电镜图片。
图9为纯聚偏氟乙烯以及含量为1wt%的纯碳纳米管、对比例1中聚甲基丙烯酸甲酯包覆碳纳米管填充聚偏氟乙烯复合材料的热失重曲线。
具体实施方式
本发明首先提供一种丙烯酸酯类聚合物包覆的碳纳米管的制备方法,该方法包括:
步骤一:将碳纳米管和多巴胺加入到tris-hcl水溶液中搅拌,所述的tris-hcl水溶液的ph为8-10,所述的搅拌温度优选为室温,反应时间优选为12-20h,得到的产物优选先离心分离,沉淀物用去离子水洗涤多次后真空干燥,得到聚多巴胺包覆的碳纳米管;
所述的碳纳米管和多巴胺的质量比优选为1:(1-10);更优选为1:2;
所述的tris-hcl水溶液的配制为:将三羟甲基氨基甲烷(tris)加入到去离子水中,室温下磁力搅拌使之完全溶解,向得到的溶液中加入盐酸溶液(hcl)调节ph,得到tris-hcl水溶液。
步骤二:将步骤一得到的聚多巴胺包覆的碳纳米管分散在溶剂中,所述的溶剂优选为二氯甲烷,然后在冰浴下加入到反应容器中,在反应容器中加入三乙胺搅拌,所述的搅拌时间优选为20-60min,更优选为30min,n2保护,然后再滴入含有2-溴异丁酰溴的二氯甲烷溶液搅拌,所述的滴加时间优选为2-3h,所述的搅拌时间优选为1-3h,更优选为3h,将温度升至室温继续反应,所述的反应时间优选为18-24h,更优选为20h,将得到的产物优选经过离心分离,所述的转速优选为为8000-10000rpm,得到的沉淀物再次分散到二氯甲烷中,离心分离,重复多次直至除去未参加反应的2-溴异丁酰溴,最后将得到的产物真空干燥,所述的干燥温度优选为60-80℃,干燥时间优选为24-48h,得到溴化碳纳米管;
所述的聚多巴胺包覆的碳纳米管和含有2-溴异丁酰溴的二氯甲烷溶液中的2-溴异丁酰溴的质量比优选为1:(5-10),更优选为1:8.5,所述的聚多巴胺包覆的碳纳米管的质量g:三乙胺的体积ml优选为1:(5-10),更优选为1:5;
步骤三:将步骤二得到的溴化碳纳米管作为引发剂、催化剂、配体和丙烯酸酯类单体加入到反应溶剂中,得到的混合物在冰浴下超声分散均匀后,对反应瓶抽真空-通氮气,完全除去瓶中氧气,随后在50-80℃下进行atrp聚合,所述的聚合温度优选为60℃,所述的聚合时间优选为6-24小时,更优选为24h,将得到的产物优选在8000-10000rpm下离心分离,得到的沉淀物再次分散到溶剂中,随后离心分离,重复多次,直至除去残留的催化剂、配体和未反应的单体,最后,将得到的产物真空干燥,所述的干燥温度优选为60-80℃,干燥时间优选为24-48h,在得到丙烯酸酯类聚合物包覆的碳纳米管,所述引发剂、催化剂、配体和丙烯酸酯类单体的质量比优选为1:1:1.2:(20-40)。
按照本发明,所述催化剂优选为氯化亚铜(cucl)或溴化亚铜(cubr),所述配体优选为2-联吡啶(bpy)、n,n,n',n,'n”-五甲基二亚乙基三胺(pmdeta)或四甲基乙二胺(tmeda),更优选为n,n,n',n,'n”-五甲基二亚乙基三胺(pmdeta),所述丙烯酸酯类单体优选为甲基丙烯酸甲酯、甲基丙烯酸乙酯、甲基丙烯酸丁酯、甲基丙烯酸缩水甘油酯、丙烯酸甲酯、丙烯酸乙酯或丙烯酸丁酯,更优选为甲基丙烯酸甲酯,反应溶剂优选为n,n-二甲基甲酰胺(dmf)。
本发明还提供上述制备方法得到的丙烯酸酯类聚合物包覆的碳纳米管。
本发明利用多巴胺在碱性水溶液中的自聚合性而粘附到碳纳米管表面,粘合力强,整个过程不需要对碳纳米管进行酸化等处理,保留了碳纳米管独特的结构和优异的性能,并且不使用溶剂,是一种环境友好型的碳纳米管表面改性方法。其次,本发明中聚多巴胺起到了桥梁的作用,灵活的运用了聚多巴胺层上存在的大量的氨基和羟基,为接上原子转移自由基聚合引发剂2-溴异丁酰溴提供了反应位点,进而引发丙烯酸酯类单体进行原位自由基聚合,实现对碳纳米管的有效包覆。本发明聚合物包覆后的碳纳米管可以实现填料在聚合物树脂基体中的均匀分散,改善填料与基体之间的界面结合性能。
下面结合附图和实施例对本发明做进一步详细说明。
实施例和对比例中所用的化学试剂:多巴胺(da),99%,sigma-aldrich公司;三羟甲基氨基甲烷(tris),99%,aldrich公司;氯化亚铜(cucl),99%,aldrich公司;溴化亚铜(cubr),99%,aldrich公司;2-溴异丁酰溴,99%,aldrich公司;n,n,n',n,'n”-五甲基二亚乙基三胺(pmdeta),99%,aldrich公司;甲基丙烯酸甲酯(mma)有吉化公司提供,使用前通过碱性氧化铝层析柱后保存;n,n-二甲基甲酰胺(dmf),甲醇,苯甲醚,光谱纯,99.9%,aldrich公司;二氯甲烷,三乙胺均为分析纯,北京化学试剂公司提供。碳纳米管购买于中科院成都有机化学研究所。聚偏氟乙烯(solef1015/1001,france)购买于东莞文锦新材料有限公司。
实施例1
(1)聚多巴胺包覆碳纳米管的制备
(a)将0.61g三羟甲基氨基甲烷(tris)加入到500ml去离子水中,室温下磁力搅拌使之完全溶解。向得到的溶液中加入1.5ml浓度1mol/l的盐酸溶液(hcl)调节ph,得到10mm、ph为8.5的tris-hcl水溶液。
(b)取步骤(a)中tris-hcl水溶液200ml,向其中加入0.2g原始碳纳米管和0.4g多巴胺,将得到的混合物在室温下剧烈搅拌12个小时。
(c)将步骤(b)所得到的产物在10000rpm下离心分离,将上层清液倒掉后,得到的沉淀物用去离子水洗涤,随后离心分离,这个过程重复多次以除去未粘附在碳纳米管表面的多巴胺单体。
(d)将步骤(c)得到的产物真空干燥48小时,即得到聚多巴胺包覆碳纳米管。
(2)溴化碳纳米管的制备
(a)取(1)中得到的产物0.2g加入到100ml二氯甲烷中,冰浴下超声分散30分钟后,随后转移到250ml的单口圆底烧瓶中并向烧瓶中加入1ml三乙胺,继续在冰浴下搅拌30分钟后,向上述溶液中逐滴加入含有1.7g2-溴异丁酰溴的50ml二氯甲烷溶液,n2保护,滴加时间为2.5h,滴加完成后继续在冰浴中搅拌3小时,随后在室温下继续反应20小时。
(b)将步骤(a)所得到的产物在10000rpm下离心分离,将上层黄色液体倒掉后,得到的沉淀物再次分散到二氯甲烷中,随后离心分离。这个过程重复多次以除去未参加反应的2-溴异丁酰溴。
(c)将步骤(b)得到的产物在60摄氏度下真空干燥24小时,得到原子转移自由基聚合的引发剂溴化碳管。
(3)原位引发丙烯酸酯类单体进行原子转移自由基聚合
取0.25g溴化碳纳米管加入到100mln,n-二甲基甲酰胺中,超声分散均匀后,向反应瓶中加入5g甲基丙烯酸甲酯,0.25gcubr,用橡皮塞将反应瓶密封,对反应瓶抽真空-通氮气,重复五次以完全除去瓶中氧气,随后用注射器向瓶中加入0.3gpmdeta,对反应瓶抽真空之后,聚合反应在60℃下进行,聚合时间为24小时。聚合完成之后将得到的产物离心分离,沉淀物随后分散到n,n-二甲基甲酰胺中,离心分离,整个过程重复多次以除去残留的配体、催化剂和未反应的单体,最后产物在80℃下真空干燥24小时,得到聚甲基丙烯酸甲酯包覆的碳纳米管。
将实施例1制备得到的聚甲基丙烯酸甲酯包覆的碳纳米管作为填料与聚偏氟乙烯树脂(pvdf)进行溶液共混,所述的聚甲基丙烯酸甲酯包覆的碳纳米管与聚偏氟乙烯树脂的质量比为1:99,得到聚甲基丙烯酸甲酯包覆碳纳米管填充聚偏氟乙烯纳米复合材料,并对材料进行形貌观察和耐热性分析。
图1为原始碳纳米管、聚多巴胺包覆的碳纳米管、溴化碳纳米管、聚甲基丙烯酸甲酯包覆碳纳米管以及聚甲基丙烯酸甲酯的傅里叶红外线光谱对比图。与纯的碳纳米管红外谱图相比,聚多巴胺改性碳纳米管的红外谱图在3395cm-1处吸收峰明显增强,这是聚多巴胺上-oh的伸缩振动峰,表面聚多巴胺成功的粘附在碳纳米管表面。从溴化碳纳米管的红外谱图中可以看出在1460cm-1位置处出现一个新的吸收峰,这归因于2-溴异丁酰溴与聚多巴胺表面的羟基反应后形成的酰胺键的吸收峰,表明atrp引发剂成功的固定到碳纳米管表面。而聚甲基丙烯酸甲酯包覆碳纳米管的红外谱图中可以明显的观察到聚甲基丙烯酸甲酯的酯基基团在1730cm-1位置处的吸收峰,表明聚甲基丙烯酸甲酯成功的包覆在碳纳米管表面。
图2为原始碳纳米管、聚多巴胺包覆的碳纳米管、溴化碳纳米管、聚甲基丙烯酸甲酯包覆碳纳米管以及聚甲基丙烯酸甲酯的热失重曲线对比图。各组分曲线在600℃时的热失重量依次增加,其中聚甲基丙烯酸甲酯包覆碳纳米管的热失重量达到58wt%,表明聚甲基丙烯酸甲酯成功的包覆在碳纳米管表面。
图3为原始碳纳米管(图a),聚多巴胺改性碳纳米管(图b)以及聚甲基丙烯酸甲酯包覆碳纳米管(图c)的透射电镜图片,从图中可以看出,与纯的碳纳米管相比,聚多巴胺改性后的碳纳米管长度没有明显变短,并且表面出现不规则的厚度约为4nm的粗糙壳层,表明聚多巴胺可以在不破坏碳纳米管结构的前提下成功的粘附到碳纳米管表面,无损改性成功。从聚甲基丙烯酸甲酯包覆碳纳米管的透射电镜图片可以看出大多数的碳纳米管表面都被聚合物包覆,碳纳米管表面出现均匀、光滑的壳层,且厚度进一步增加,表明聚甲基丙烯酸甲酯成功的包覆在聚多巴胺改性的碳纳米管上。
图4为1wt%纯碳纳米管(图a)、聚甲基丙烯酸甲酯包覆碳纳米管分散在聚偏氟乙烯(图b)基体中断面扫描电镜图片。从图4a中可以看出,原始的碳纳米管在pvdf基体中有明显的团聚和界面剥离现象发生,这主要是由于未经处理的碳纳米管与pvdf之间的相容性较差。而聚甲基丙烯酸甲酯包覆的碳纳米管可以均匀的分散在pvdf基体中(图4b),没有明显的团聚和剥离现象发生,这表明聚甲基丙烯酸甲酯包覆后的碳纳米管与pvdf之间的相容性良好,进而促进了碳纳米管在聚合物基体中的均匀分散。
图5为聚偏氟乙烯以及含量为1wt%的纯碳纳米管、聚甲基丙烯酸甲酯包覆碳纳米管填充聚偏氟乙烯复合材料的热失重曲线。从图中可以看出,与纯聚偏氟乙烯和纯碳纳米管填充聚偏氟乙烯复合材料相比,聚甲基丙烯酸甲酯包覆碳纳米管填充聚偏氟乙烯纳米复合材料在高温下呈现出增加的热稳定性,表明包覆碳纳米管对提高碳纳米管在基体中的分散性以及所得复合材料的综合性能具有重要意义。
实施例2
(1)聚多巴胺包覆碳纳米管的制备
(a)将0.61g三羟甲基氨基甲烷(tris)加入到500ml去离子水中,室温下磁力搅拌使之完全溶解。向得到的溶液中加入1.5ml浓度为1mol/l的盐酸溶液(hcl)调节ph,得到10mm、ph为8.5的tris-hcl水溶液。
(b)取步骤(a)中tris-hcl水溶液200ml,向其中加入0.2g原始碳纳米管和0.4g多巴胺,将得到的混合物在室温下剧烈搅拌12个小时。
(c)将步骤(b)所得到的产物在10000rpm下离心分离,将上层清液倒掉后,得到的沉淀物用去离子水洗涤,随后离心分离。这个过程重复多次以除去未粘附在碳纳米管表面的多巴胺单体。
(d)将步骤(c)得到的产物真空干燥48小时,即得到聚多巴胺包覆碳纳米管。
(2)溴化碳纳米管的制备
(a)取(1)中得到的产物0.2g加入到100ml二氯甲烷中,冰浴下超声分散30分钟后,随后转移到250ml的单口圆底烧瓶中并向烧瓶中加入1ml三乙胺,继续在冰浴下搅拌30分钟后,向上述溶液中逐滴加入含有1.7g2-溴异丁酰溴的50ml二氯甲烷溶液,n2保护,滴加时间为2.5h,滴加完成后继续在冰浴中搅拌3小时,随后在室温下继续反应20小时。
(b)将步骤(a)所得到的产物在10000rpm下离心分离,将上层黄色液体倒掉后,得到的沉淀物再次分散到二氯甲烷中,随后离心分离。这个过程重复多次以除去未参加反应的2-溴异丁酰溴。
(c)将步骤(b)得到的产物在60摄氏度下真空干燥24小时,得到原子转移自由基聚合的引发剂溴化碳纳米管。
(3)原位引发丙烯酸酯类单体进行原子转移自由基聚合
取0.25g溴化碳纳米管加入到100mln,n-二甲基甲酰胺中,超声分散均匀后,向反应瓶中加入10g甲基丙烯酸甲酯,0.25gcucl,用橡皮塞将反应瓶密封,对反应瓶抽真空-通氮气,重复五次以完全除去瓶中氧气,随后用注射器向瓶中加入0.3gpmdeta,对反应瓶抽真空之后,聚合反应在60℃下进行,聚合时间为24小时。聚合完成之后将得到的产物离心分离,沉淀物随后分散到n,n-二甲基甲酰胺中,离心分离,整个过程重复多次以除去残留的配体、催化剂和未反应的单体,最后产物在80℃下真空干燥24小时,得到聚甲基丙烯酸甲酯包覆的碳纳米管。
对实施例2制备得到上述材料进行热失重测试,聚甲基丙烯酸甲酯包覆碳纳米管在600℃时失重量为52.4wt%,表明聚甲基丙烯酸甲酯成功的包覆到碳纳米管表面上。
对比例1
(1)聚多巴胺包覆碳纳米管的制备
(a)将0.61g三羟甲基氨基甲烷(tris)加入到500ml去离子水中,室温下磁力搅拌使之完全溶解。向得到的溶液中加入1.5ml浓度为1mol/l的盐酸溶液(hcl)调节ph,得到10mm、ph为8.5的tris-hcl水溶液。
(b)取步骤(a)中tris-hcl水溶液200ml,向其中加入0.2g原始碳纳米管和0.4g多巴胺,将得到的混合物在室温下剧烈搅拌12个小时。
(c)将步骤(b)所得到的产物在10000rpm下离心分离,将上层清液倒掉后,得到的沉淀物用去离子水洗涤,随后离心分离。这个过程重复多次以除去未粘附在碳纳米管表面的多巴胺单体。
(d)将步骤(c)得到的产物真空干燥48小时,即得到聚多巴胺包覆碳纳米管。
(2)溴化碳纳米管的制备
(a)取(1)中得到的产物0.2g加入到100ml二氯甲烷中,冰浴下超声分散30分钟后,随后转移到250ml的单口圆底烧瓶中并向烧瓶中加入1ml三乙胺,继续在冰浴下搅拌30分钟后,向上述溶液中逐滴加入含有1.7g2-溴异丁酰溴的50ml二氯甲烷溶液,n2保护,滴加时间为2.5h,滴加完成后继续在冰浴中搅拌3小时,随后在室温下继续反应20小时。
(b)将步骤(a)所得到的产物在10000rpm下离心分离,将上层黄色液体倒掉后,得到的沉淀物再次分散到二氯甲烷中,随后离心分离。这个过程重复多次以除去未参加反应的2-溴异丁酰溴。
(c)将步骤(b)得到的产物在60摄氏度下真空干燥24小时,得到原子转移自由基聚合的引发剂溴化碳纳米管。
(3)原位引发丙烯酸酯类单体进行原子转移自由基聚合
取0.25g溴化碳纳米管加入到100ml苯甲醚中,超声分散均匀后,向反应瓶中加入10g甲基丙烯酸甲酯,0.25gcubr,用橡皮塞将反应瓶密封,对反应瓶抽真空-通氮气,重复五次以完全除去瓶中氧气,随后用注射器向瓶中加入0.3gpmdeta,对反应瓶抽真空之后,聚合反应在60℃下进行,聚合时间为24小时。聚合完成之后将得到的产物离心分离,沉淀物随后分散到n,n-二甲基甲酰胺中,离心分离,整个过程重复多次以除去残留的配体、催化剂和未反应的单体,最后产物在80℃下真空干燥24小时,得到聚甲基丙烯酸甲酯包覆的碳纳米管。
将对比例1制备得到的聚甲基丙烯酸甲酯包覆的碳纳米管作为填料与聚偏氟乙烯树脂(pvdf)进行溶液共混,所述的聚甲基丙烯酸甲酯包覆的碳纳米管与聚偏氟乙烯树脂的质量比为1:99,得到聚甲基丙烯酸甲酯包覆碳纳米管填充聚偏氟乙烯纳米复合材料,并对材料进行形貌观察和耐热性分析。
图6为原始碳纳米管、聚多巴胺包覆的碳纳米管、溴化碳纳米管、对比例1中聚甲基丙烯酸甲酯包覆碳纳米管以及聚甲基丙烯酸甲酯的热失重曲线对比图。从图中可以看出,与聚多巴胺包覆碳纳米管相比,对比例1中聚甲基丙烯酸甲酯包覆碳纳米管在600℃时失重量有少量增多,表明只有少量聚甲基丙烯酸甲酯包覆到碳纳米管上。
图7为原始碳纳米管(图a),聚多巴胺改性碳纳米管(图b)以及对比例1中聚甲基丙烯酸甲酯包覆碳纳米管(图c)的透射电镜图片。从图中可以看出,与聚多巴胺改性后的碳纳米管相比,对比例1中聚甲基丙烯酸甲酯包覆碳纳米管厚度没有明显增加,表明包覆效果不明显。
图8为1wt%纯碳纳米管、对比例1中聚甲基丙烯酸甲酯包覆碳纳米管分散在聚偏氟乙烯基体中断面扫描电镜图片。从图中可以看出纯碳纳米管在聚偏氟乙烯基体中出现明显的团聚和剥离现象,如图8a所示。而对比例1中聚甲基丙烯酸甲酯包覆的碳纳米管在pvdf基体中没有明显的剥离出现,但可以观察到少量的聚集(图8b),这表明对比例1中包覆少量聚甲基丙烯酸甲酯的碳纳米管与pvdf基体的相容性比实例1差,导致对比例1中聚甲基丙烯酸甲酯包覆碳纳米管在基体中分散不均。
图9为纯聚偏氟乙烯以及含量为1wt%的纯碳纳米管、对比例1中聚甲基丙烯酸甲酯包覆碳纳米管填充聚偏氟乙烯复合材料的热失重曲线。从图中可以看出,与纯聚偏氟乙烯相比,纯碳纳米管填充聚偏氟乙烯复合材料和对比例1中聚甲基丙烯酸甲酯包覆碳纳米管填充聚偏氟乙烯纳米复合材料的热稳定性有所增加,但纯碳纳米管填充的复合材料与对比例1中聚甲基丙烯酸甲酯包覆碳纳米管填充聚偏氟乙烯复合材料在高温下的热稳定性相差不大,表明包覆较少聚合物的碳纳米管对复合材料的增强作用较弱。
对比例2
(1)聚多巴胺包覆碳纳米管的制备
(a)将0.61g三羟甲基氨基甲烷(tris)加入到500ml去离子水中,室温下磁力搅拌使之完全溶解。向得到的溶液中加入1.5ml浓度为1mol/l的盐酸溶液(hcl)调节ph,得到10mm、ph为8.5的tris-hcl水溶液。
(b)取步骤(a)中tris-hcl水溶液200ml,向其中加入0.2g原始碳纳米管和0.4g多巴胺,将得到的混合物在室温下剧烈搅拌12个小时。
(c)将步骤(b)所得到的产物在10000rpm下离心分离,将上层清液倒掉后,得到的沉淀物用去离子水洗涤,随后离心分离。这个过程重复多次以除去未粘附在碳纳米管表面的多巴胺单体。
(d)将步骤(c)得到的产物真空干燥48小时,即得到聚多巴胺包覆碳纳米管。
(2)溴化碳纳米管的制备
(a)取(1)中得到的产物0.2g加入到100ml二氯甲烷中,冰浴下超声分散30分钟后,随后转移到250ml的单口圆底烧瓶中并向烧瓶中加入1ml三乙胺,继续在冰浴下搅拌30分钟后,向上述溶液中逐滴加入含有1.7g2-溴异丁酰溴的50ml二氯甲烷溶液,n2保护,滴加时间为2.5h,滴加完成后继续在冰浴中搅拌3小时,随后在室温下继续反应20小时。
(b)将步骤(a)所得到的产物在10000rpm下离心分离,将上层黄色液体倒掉后,得到的沉淀物再次分散到二氯甲烷中,随后离心分离。这个过程重复多次以除去未参加反应的2-溴异丁酰溴。
(c)将步骤(b)得到的产物在60摄氏度下真空干燥24小时,得到原子转移自由基聚合的引发剂溴化碳纳米管。
(3)原位引发丙烯酸酯类单体进行原子转移自由基聚合
取0.25g溴化碳纳米管加入到100ml甲醇中,超声分散均匀后,向反应瓶中加入10g甲基丙烯酸甲酯,0.25gcubr,用橡皮塞将反应瓶密封,对反应瓶抽真空-通氮气,重复五次以完全除去瓶中氧气,随后用注射器向瓶中加入0.3gpmdeta,对反应瓶抽真空之后,聚合反应在60℃下进行,聚合时间为24小时。聚合完成之后将得到的产物离心分离,沉淀物随后分散到n,n-二甲基甲酰胺中,离心分离,整个过程重复多次以除去残留的配体、催化剂和未反应的单体,最后产物在80℃下真空干燥24小时,得到聚甲基丙烯酸甲酯包覆的碳纳米管。
将对比例2中制备的纳米材料进行热失重测试,聚甲基丙烯酸甲酯包覆碳纳米管在600℃时失重量为21wt%,表明少量聚甲基丙烯酸甲酯包覆到碳纳米管表面上。
从本发明的实施例和对比例可以看出,步骤(3)中用于分散溴化碳纳米管和溶解配体以及催化剂的溶剂,也是实现包覆效果好差的关键因素,当用对比例的苯甲醚或甲醇溶剂时,配体及催化剂在其中的溶解效果不是很好,导致催化剂不能有效的夺取引发剂溴化碳纳米管中的溴原子而引发聚合反应,这在一定程度上减弱了催化剂和配体的作用,导致只有少量聚丙烯酸酯类聚合物接枝到溴化碳纳米管上。
- 还没有人留言评论。精彩留言会获得点赞!