基于炔-胺点击聚合的细胞内聚合方法与流程
本发明涉及高分子化学和化学生物学领域,具体涉及一种基于炔-胺点击聚合的细胞内聚合方法。
背景技术:
在生理环境中,可高效进行的化学反应将成为探索生命活动的有力工具。聚合物体系相对于小分子具有结构易修饰、协同放大效应和不易被细胞外排等优点,因此,聚合物体系在诊断、成像和治疗领域具有更好的应用前景。基于小分子的细胞内反应目前研究较多,且取得了良好的成果,目前最经典的一类反应为生物正交反应,包括点击化学、staudinger连接、quadricylane连接、diels-alder反应等,已经被应用于细胞成像、示踪、疾病治疗、基因调控和生物大分子标记等。但是细胞内的聚合反应报道很少,主要是因为引入金属催化剂带来的细胞毒性和反应基团活性过高导致结构不稳定等问题,导致很少有聚合反应能够在生理环境中进行。为了解决细胞毒性问题,最近发展了基于生物酶的细胞内聚合体系用于原位制备寡聚物/聚合物。但是荧光标记以及反应基团的引入同样限制了体系的进一步发展。因此,发展细胞内聚合需要更简单、高效的反应体系。
2014年,唐本忠院士课题在扩展基于炔类单体的点击聚合过程中,建立了一种无需催化剂、自发进行的巯基-炔点击聚合反应。芳香双炔单体与双巯基单体等当量混合,于30℃反应2小时就能得到溶解性好、分子量高(重均分子量可达8.52×104)的聚合物。该自发进行的点击聚合不仅具有良好的区域选择性,并且还具有很好的官能团容忍性(macromalecules,2014,47:1325-1333)。
技术实现要素:
本发明的目的在于提供一种基于炔-胺点击聚合的细胞内聚合方法。该方法成功在细胞内聚合完成,反应简单、高效、条件温和。
本发明的目的通过下述技术方案实现:
一种基于炔-胺点击聚合的细胞内聚合方法,包括以下步骤:
(1)在细胞培养体系中,将二元胺基化合物与细胞共培养;
(2)去除未与细胞作用的二元胺基化合物,加入二元炔基化合物与细胞共培养;
(3)去除未与细胞作用的二元炔基化合物,加入培养基继续培养,获得细胞内聚合物。
所述二元炔基化合物的结构为式(ⅱ)或(ⅲ):
所述的二元胺基化合物的结构为式(ⅳ):
细胞内聚合物的结构为式(ⅰ):
式(ⅰ)~(ⅳ)中,n为1~1000的整数,r1,r3为相同或不同的有机基团,r2,r4为氢原子或者有机基团。
作为优选,式(ⅰ)~(ⅳ)中,r1,r3选自(1)~(18)中的任意一种;r2,r4选自氢原子或(19)~(22)中的任意一种;
其中,m、h、k为1~100整数;x选自n、p、o、s或si元素;*表示取代位置。
所述细胞培养体系为磷酸缓冲盐溶液、hank’s缓冲液(无机盐溶液和平衡盐溶液)、tris缓冲液(三(羟甲基)氨基甲烷缓冲液)、hepes缓冲液(4-羟乙基哌嗪乙磺酸缓冲液)、dmem细胞培养基(dulbecco's改良培养基)中的至少一种,优选为磷酸缓冲盐溶液或hank’s缓冲液,此时得到的聚烯胺类化合物分子量较高。
步骤(1)~(2)中所述共培养的温度各自独立地为4~37℃。
步骤(3)中所述继续培养的温度为4~37℃。
步骤(1)~(2)中所述共培养的时间各自独立地为1~300min,优选为15~30min。
步骤(1)中所述二元胺基化合物在细胞培养体系中的浓度为0.0001~100mmol/l,优选为1~1000um。步骤(1)中所述二元胺基化合物与步骤(2)中二元炔基化合物的摩尔比为1:1。
步骤(2)中所述二元炔基化合物以溶液的形式加入,溶剂为细胞培养体系;
步骤(3)中所述继续培养的时间为10~600min,优选为120~240min。
步骤(2)中所述去除未与细胞作用的二元胺基化合物是指采用pbs进行洗涤;
步骤(3)中所述去除未与细胞作用的二元炔基化合物是指采用pbs进行洗涤;
步骤(3)中所述培养基为含10%fbs(胎牛血清)的dmem培养基。
步骤(1)~(3)中所述细胞可为肿瘤细胞,为肿瘤细胞时,本发明的方法具有抗肿瘤的效果。
一种基于炔-胺点击聚合的细胞内荧光成像方法,包括以下步骤:
(1)在细胞培养体系中,加入二元胺基化合物与细胞共培养;
(2)去除未与细胞作用的二元胺基化合物,加入二元炔基化合物与细胞共培养;
(3)去除未与细胞作用的二元炔基化合物,加入培养基继续培养,激光共聚焦成像,实现细胞内荧光成像。激发光源为405nm。
所述细胞中生物大分子包括含有胺基的生物大分子。步骤(1)~(3)中条件与细胞内聚合方法相同。
本发明的细胞内荧光成像是由于二元炔基化合物和二元胺基化合物在细胞内进行聚合得到的聚合物末端的炔基能够进一步与生物大分子上的胺基反应,实现对生物大分子的原位标记,且该细胞内聚合反应能够锚定小分子荧光材料,导致荧光信号“点亮”,实现细胞的荧光成像。
另外,本发明的基于炔-胺点击聚合的细胞内聚合方法中当细胞为肿瘤细胞时,二元炔基化合物和二元胺基化合物在细胞内进行聚合制备聚合物的过程中会破坏肿瘤细胞内微管结构,导致肿瘤细胞的坏死,实现肿瘤细胞杀伤。若是将二元胺基化合物与二元炔基化合物反应制得的聚烯胺类化合物与细胞共培养,聚合物不能与细胞相互作用,且不能杀死肿瘤细胞。
与现有技术相比,本发明具有如下特点:
1、本发明直接利用炔类单体和胺在细胞内进行点击聚氢胺化反应成功在细胞内合成了聚烯胺类化合物,实现了细胞内聚合。
2、该细胞内聚合方法,条件温和,反应迅速,操作简单。
3、该细胞内聚合方法,无需通过基因工程,代谢和化学修饰手段引入反应官能团,能够原位与细胞内生物大分子上的氨基反应,且能够通过聚合的手段锚定小分子荧光材料,使小分子不易被细胞外排,并通过抑制分子内运动导致荧光“点亮”,具有高的信噪比,实现细胞的高灵敏荧光成像。
4、该细胞内聚合方法,在二元炔基化合物和二元胺基化合物细胞毒性不明显的情况下,在细胞内原位生成的聚合物能够破坏肿瘤细胞的微管结构,导致肿瘤细胞的坏死,实现肿瘤细胞的杀伤,具有抗肿瘤的应用前景。
附图说明
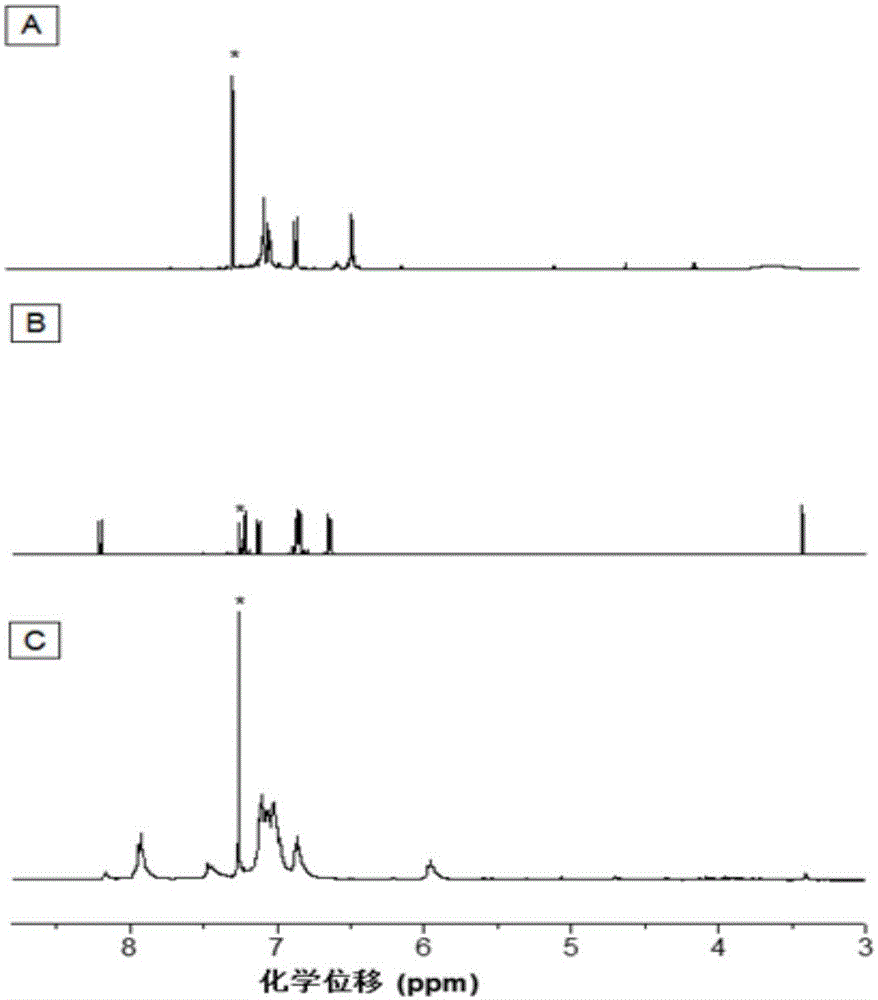
图1为对比例1中单体m1(a),单体m2(b)和制备的聚烯胺类化合物p1(c)在cdcl3中核磁共振氢谱对比图(*代表溶剂峰);
图2为对比例1中单体m1(a),单体m2(b)在cdcl3中核磁共振碳谱和制备的聚烯胺类化合物p1(c)在氘代四氢呋喃中核磁共振碳谱对比图(*代表溶剂峰);
图3为不同分子量的聚合物光谱图,其中分子量为7129的为细胞内聚合物的发光光谱;
图4为实施例2中hela细胞用单体m1(a),单体m2(b)和细胞内聚合(c)分别处理的激光共聚焦显微镜成像图;
图5为实施例2中单体m1和单体m2聚合得到的聚合物与hela细胞作用(a),单体m1与单体m2同时与细胞相互作用(b),单体m1与单体m2先后与细胞相互作用(即细胞内聚合)(c)的激光共聚焦显微镜成像图;
图6为实施例2中单体m1和m2先后与细胞(hek293,hcc827,mcf7,死hela细胞)作用后的激光共聚焦显微镜成像图;
图7为实施例3单体m1(a),单体m2(b)和细胞内聚合(c)对hela细胞毒性测试结果图;
图8为实施例3中hela细胞分别用单体m1,单体m2和细胞内聚合处理后,与肌动蛋白共染的激光共聚焦显微镜成像图;
图9为实施例3中hela细胞分别用单体m1,单体m2和细胞内聚合处理后,与微管蛋白共染的激光共聚焦显微镜成像图;
图10为实施例3中空白组和细胞内聚合(即实验组)的hela细胞扫描电镜表征图。
具体实施方式
下面结合实施例对本发明进行具体地描述,但本发明的保护范围不限于以下实施例。
对比例1
一种聚烯胺类化合物,其结构式如p1所示:
所述聚烯胺类化合物通过炔类单体与二元胺经点击氢胺化聚合反应进行制备,反应方程式如式(一):
其中,单体m1的合成方法可按照已公开文献中(macromolecules1997,30:3547-3552)的合成方法合成;单体m2的合成按照反应方程式如式(二):
所述的聚烯胺类化合物的制备步骤如下:
36.2mg(0.1mmol)单体m1和27.4mg(0.1mmol)单体m2分别溶解在50μl四氢呋喃中,并依次加入装有4.9毫升水的10毫升聚合管中,25℃搅拌3小时,然后离心收集沉淀。将沉淀用四氢呋喃溶解后,将得到的聚合物溶液滴加到剧烈搅拌的正己烷中,然后静置,过滤,干燥,得到目标聚合物p1。经测定分析,最终产物聚烯胺类化合物p1的产率为97%,重均分子量为11057,分子量分布为1.27。该聚合物与其相应单体的核磁共振氢谱对比图见图1,氨基氢和炔基氢的消失说明聚合反应成功进行。该聚合物与其对应的单体核磁共振碳谱见图2。此外,还测定了单体m1和聚烯胺类化合物p1的光谱,显示单体m1聚合成聚烯胺类化合物p1后无论吸收光谱还是荧光发射都发生红移。通过测试不同四氢呋喃/水的混合溶解中的荧光光谱,测试结果显示聚烯胺类化合物p1具有明显的聚集诱导发光现象。
实施例1
一种基于炔-胺点击聚合的细胞内的聚合方法,包括以下步骤:
(1)在1ml的细胞培养体系(pbs溶液)中,加入二元胺基化合物(即单体m1)与hela细胞37℃共培养20min;二元胺基化合物在细胞培养体系中的浓度为10μm;
(2)用pbs洗3次后去除未与细胞作用的二元胺基化合物,加入二元炔基化合物(即单体m2)与hela细胞37℃共培养20min;二元炔基化合物与二元胺基化合物的摩尔比为1:1;二元炔基化合物在细胞培养体系中的浓度为10μm;
(3)用pbs洗涤或者吸出单体溶液(去除未与细胞作用的二元炔基化合物),加入1ml的dmem(10%胎牛血清)培养基培养3小时,获得细胞内聚合物;
二元胺基化合物为
将上述培养完后的细胞,先用1ml水裂解后,离心获得细胞碎片的沉淀。加入1ml胰酶水解蛋白质,继续离心收集沉淀。将得到的沉淀用1ml四氢呋喃溶解提取制备的聚合物,将得到的聚合物溶液滴加到剧烈搅拌的正己烷中,然后静置,过滤,干燥,测得聚合物的重均分子量7129,分子量分布为1.65。另外,单纯只用单体(二元胺基化合物和二元炔基化合物)单独作用的细胞中检测不到分子量信号(即只用二元胺化合物或只用二元炔基化合物与细胞培养,所获得产物中检测不到分子量信号)。上述实验结果证明单体m1和单体m2能在细胞内发生聚合。
在提取聚合物的过程中,大部分聚合物依然与细胞依然连接在一起,无法一起溶解。并且当选择含有大量氨基酸的培养基与细胞培养时,细胞内荧光信号会明显降低,说明炔基能够与蛋白质上的氨基相互作用。说明聚合物能够与细胞内生物大分子共价连接,实现对生物大分子的原位标记。
将本实施例通过细胞内聚合方法得到的聚合物以及不同分子量的聚合物进行光谱测试,测试结果如图3所示。图3为不同分子量的聚合物光谱图,其中分子量为7129的为细胞内聚合物的发光光谱;其他几种分子量的聚合物是通过对比例1的方法制备得到(调整单体的用量以及反应时间即可获得不同分子量的聚合物)。从图3侧面反映出细胞内聚合反应确实已经发生,且分子量大于2300,小于11000)。
实施例2
细胞内聚合用于生物大分子标记和细胞成像:(单体m1和单体m2同对比例1中的单体m1和单体m2)
细胞内聚合:将hela细胞(购自协和细胞资源中心)培养24小时后,与10μm单体m1(二元胺基化合物)的pbs溶液37℃培养20分钟,用pbs洗3次后,继续与10μm单体m2(二元炔基化合物)的pbs溶液37℃培养20分钟,吸走单体溶液,加入1mldmem(10wt%胎牛血清)培养基培养3小时,用激光共聚焦显微镜成像。
单体单独与细胞作用:将单体m1和单体m2(10um)分别单独与hela细胞(细胞培养24h后)37℃培养20分钟,去除单体溶液,加入dmem(10%胎牛血清)培养基继续培养180分钟,用激光共聚焦成像。
图4为实施例2中hela细胞用单体m1(a),单体m2(b)和细胞内聚合(c)分别处理的激光共聚焦显微镜成像图。单体单独处理的细胞检测不到荧光信号,而只有将单体m1和单体m2先后与细胞作用,在细胞内聚合才能检测到荧光信号。
为了证明聚合反应确实在细胞内进行,而非生成的聚合物与细胞相互作用形成的实验结果。现将细胞分别与已经制得的聚合物p1及单体m1和单体m2同时加入磷酸缓冲液中,37℃作用180分钟后,用激光共聚焦显微镜成像。结果见图5。图5为实施例2中单体m1和单体m2聚合得到的聚合物与hela细胞作用(a),单体m1与单体m2同时与细胞相互作用(b),单体m1与单体m2先后与细胞相互作用(即细胞内聚合)(c)的激光共聚焦显微镜成像图。发现,聚合物p1与细胞作用以及单体m1和单体m2同时与细胞作用都没有在细胞内观察到荧光信号。而单体m1与单体m2先后与细胞相互作用(即细胞内聚合)有荧光信号,证明细胞内的荧光信号来自单体在细胞内聚合实现的。
为了验证细胞内聚合的普适性,将正常细胞hek293,正常细胞hcc827,肿瘤细胞mcf7,肿瘤细胞死的hela分别与单体(单体m1和m2先后培养)的pbs溶液共培养20分钟后,加入培养基继续培养180分钟,在共聚焦显微镜下成像,均能检测到明显的荧光信号,证明这个细胞内聚合物方法具有普适性,结果见图6。图6为实施例2中单体m1和m2先后与细胞(hek293,hcc827,mcf7,死hela细胞)作用后的激光共聚焦显微镜成像图。
实施例3
细胞内聚合对肿瘤细胞杀伤:单体m1和单体m2同对比例1中单体m1和单体m2;
将hela细胞铺在96孔板中培养24小时后,先与不同浓度单体m1(0.5,1,2,4,8,16,32,64μm)的pbs溶液作用20分钟,吸走液体后(或pbs洗涤3次),加入相应浓度单体m2的pbs溶液,培养20分钟,吸走液体,加入100μldmem(10%胎牛血清)培养基培养24小时后,加入mtt培养4小时,测570nm处的紫外吸收强度。对照组中只分别用单体m1或m2培养,其他操作一致。测试结果见图7。图7为实施例3中单体m1(a),单体m2(b)和细胞内聚合(即单体m1和单体m2先后作用细胞,在细胞内聚合)(c)对hela细胞毒性测试结果图。单体m1基本没有毒性,单体m2有一定毒性,但是当两种单体相继与细胞作用后,细胞毒性明显增加,说明细胞内聚合对肿瘤细胞具有明显的杀伤效果。
将分别与单体m1、单体m2作用的细胞以及细胞内聚合的细胞,分别与标记有红色荧光分子的肌动蛋白抗体和微管蛋白抗体作用,发现细胞内聚合会一定程度破坏肌动蛋白,且对细胞内微管蛋白的结构破坏非常明显,结果见图8和图9。图8为实施例3中hela细胞分别用单体m1,单体m2和细胞内聚合处理后,与肌动蛋白共染的激光共聚焦显微镜成像图;图9为实施例3中hela细胞分别用单体m1,单体m2和细胞内聚合处理后,与微管蛋白共染的激光共聚焦显微镜成像图。
进一步用扫描电镜表征,结果见图10。图10为实施例3中空白组和细胞内聚合(实验组)的hela细胞扫描电镜表征图。发现与空白组细胞相比,细胞结构破坏严重且释放出内含物,说明细胞因为微管蛋白结构破坏导致细胞坏死,从而实现抗肿瘤效果。
- 还没有人留言评论。精彩留言会获得点赞!