喹唑啉化合物、其制备方法和应用与流程
本发明涉及一种喹唑啉化合物、其制备方法和应用。
背景技术:
肺癌是全球头号癌症杀手,近年来发病率在全球范围内持续上升,而在中国尤其明显。据《中国肿瘤登记年报》报道,在2012年,肺癌已取代肝癌成为我国首位恶性肿瘤死亡原因,占全部恶性肿瘤死亡的22.7%。1988-2005年间我国肺癌发病率年均增长1.63%,如不及时采取有效控制措施,预计到2025年,我国将成为世界第一肺癌发病大国(郝捷,陈万青.2012中国肿瘤登记年报[m].军事医学科学出版社,2012.姚晓军,刘伦旭.肺癌的流行病学及治疗现状[j].现代肿瘤医学,2014,22(8):1982-1986.)。
在所有的肺癌患者中,非小细胞肺癌(nsclc)(包括鳞癌、腺癌、大细胞癌)患者多达85%(jemala,siegelr,xuj,etal.cancerstatistics,2010[j].ca-cancerjclin,2010,60(5):277-300.)。目前,nsclc的表皮生长因子受体(egfr)靶向治疗极具前景。egfr是原癌基因erbbl(her-l)的表达产物,是一个具有酪氨酸激酶活性的跨膜糖蛋白,研究表明许多肿瘤中都存在着egfr的表达增高或异常表达,在大多数的nsclc病例中都能够检测到egfr的过量表达,鳞癌中过量表达egfr的比例多达90%,而腺癌中这一比例为30-65%(sequistlv,lynchtj.egfrtyrosinekinaseinhibitorsinlungcancer:anevolvingstory[j].annurevmed,2008,59:429-442.dutut,michielss,fouretp,etal.differentialexpressionofbiomarkersinlungadenocarcinoma:acomparativestudybetweensmokersandnever-smokers[j].annoncoldec,2005,16(12):1906-1914.)。egfr与肿瘤细胞的增殖、新生血管生成、肿瘤侵袭、转移及预后密切相关,因此是非常有潜力的肿瘤治疗和显像靶点。
目前,最具应用前景且研究较为深入的主要是egfr酪氨酸激酶小分子抑制剂(egfr-tki)和单克隆抗体(mab)。fda批准了四个egfr-tki用于治疗nsclc,分别是:阿法替尼、吉非替尼、厄洛替尼和拉帕替尼。埃克替尼是中国自主研发的一种强效、高选择性的靶向egfr-tki,是继吉非替尼(易瑞沙)和厄洛替尼(特罗凯)之后全球第三个用于晚期肺癌的egfr酪氨酸激酶抑制剂。分别于2011年6月和2014年11月获得国家食品药品监督管理局批准适用nsclc患者二、三线用药和一线治疗,其结构如下所示。
通常,egfr的分子靶向药物不仅较常用的细胞毒性药物价格昂贵,还可能会引起皮炎、腹泻等副作用。同时,不加选择地让患者使用egfr分子靶向药物,会使得治疗的总有效率偏低。因此,为了有效的利用分子靶向药物,治疗前对egfr表达水平和突变状态以及早期疗效的评价显得十分必要。
靶向egfr的pet显像可以实现对egfr靶向药物的疗效早期精确检测,从而促进nsclc个体化egfr靶向治疗。当前报道的该类pet显像剂如下所示,然而,其中大都存在肝、肾和胃肠道等器官放射性浓度较高,对肿瘤选择性不够高的问题,且应用具有较多的局限性。f-18pet显像剂的制备,一般需要设计复杂的标记前体或是制备过程繁琐、标记率和放射化学产率较低,部分探针还存在制备过程繁琐,结构稳定性差的缺点。c-11标记的探针,因c-11的半衰期较短(20.4min),也限制了该类探针只能在有加速器的医疗机构使用。
因此,亟需开发一种新的靶向egfr的pet显像剂,用于在临床上筛选egfr分子靶向治疗的敏感人群,监测治疗效果以及评估患者预后情况,以实现对肿瘤的早期诊断、准确分期、监测复发和转移,协助确定肿瘤治疗方案,提高肿瘤治疗的效果和降低死亡率。因此,新的靶向egfr的pet显像剂的研究具有重要的现实意义和应用价值。
技术实现要素:
本发明所要解决的技术问题是克服现有的靶向egfr的pet显像剂对肿瘤选择性不够高、放射化学产率低、制备复杂繁琐的问题,而提供了一种喹唑啉化合物、其制备方法和应用。该喹唑啉化合物作为pet显像剂,结构稳定,显像分辨率高,可以精确、无创的用于egfr过量表达的肿瘤的诊断和显像。此外,其制备方法简单,反应时间短,放射性化学产率和放射化学纯度高,适合工业化生产。
本发明主要是通过以下技术方案解决上述技术问题的。
本发明提供了一种如式i所示的喹唑啉化合物或其药学上可接受的盐:
其中,r1为c2~c10直链烷基、c2~c10支链烷基、c5~c8环烷基、
x为18f或f。
当r1为c2~c10直链烷基时,所述c2~c10直链烷基优选为c2~c3直链烷基。
当r1为c2~c10支链烷基时,所述c2~c10支链烷基优选为
当r1为c5~c8环烷基时,所述c5~c8环烷基优选为
当r1为
当r1为c6~c10芳基-c1~c4烷基时,所述c6~c10芳基-c1~c4烷基优选为
当r1为c1~c4烷基-c6~c10芳基-c1~c4烷基时,所述c1~c4烷基-c6~c10芳基-c1~c4烷基优选为
在某一技术方案中,所述如式i所示的喹唑啉化合物或其药学上可接受的盐中某些基团的定义可如下所述(未定义的基团如前任一所述):
r1为c2~c10直链烷基;x为18f或f。
在某一技术方案中,所述如式i所示的喹唑啉化合物或其药学上可接受的盐中某些基团的定义可如下所述(未定义的基团如前任一所述):
r1为c2~c3直链烷基;x为18f或f。
所述如式i所示的喹唑啉化合物可为以下任一结构,
本发明提供了一种如式i所示喹唑啉化合物的制备方法,其包括以下步骤:在溶剂中,在一价铜盐的催化下,将如式b所示化合物和如式c所示化合物进行1,3-偶极环加成反应,即可;
其中,r1和x的定义如前所述。
所述制备方法中,所述溶剂可为本领域常规,例如水、叔丁醇、乙腈、四氢呋喃、dmf(n,n-二甲基甲酰胺)和dmso(二甲基亚砜)中的一种或多种。
所述制备方法中,所述化合物b在所述溶剂中的浓度可为本领域常规,例如0.1~50mmol/l,再例如2~20mmol/l。
所述制备方法中,当x为18f时,所述化合物c在所述溶剂中的浓度可为本领域常规,例如0.02~2000mci/ml,再例如1~500mci/ml,再例如10~200mci/ml。
所述制备方法中,当x为f时,所述化合物c在所述溶剂中的浓度可为本领域常规,例如1~20mol/l,再例如1~6mol/l。
所述制备方法中,所述一价铜盐可为本领域该类反应常规使用的一价铜盐。
所述制备方法中,所述一价铜盐在所述溶剂中的浓度可为本领域常规,例如0.05~2mol/l。
所述制备方法中,所述1,3-偶极环加成反应的温度可为本领域常规,例如2~110℃,再例如35~80℃。
所述制备方法中,所述1,3-偶极环加成反应的时间可为本领域常规,例如1~80min,再例如10~60min。
所述制备方法中,当x为18f时,所述如式c所示化合物为化合物c1,所述的制备方法还可进一步包括下述步骤:在有机溶剂中,将k222、k2co3和18f-与化合物d进行如下所示的亲核取代反应,即可;
其中,r2为亲核取代反应中常规的离去基团,例如-ots、-oms、-no2、i-、br-、-otf或-n+me3。
所述亲核取代反应的条件和操作均与本领域该类反应常规的条件和操作相同;所述化合物d市售可得,或可通过文献制备获得(郭飞虎;施玲丽;王妮;武明星;张勇平;杜进.p-[18f]fstc的合成和初步动物学评价,核化学与放射化学,2010,32(4):236-242;gillhs,marikj.preparationof(18)f-labeledpeptidesusingthecopper(i)-catalyzedazide-alkyne1,3-dipolarcycloaddition.natureprotocols2011;6(11):1718-1725.)。
所述制备方法中,当x为f时,所述如式c所示化合物为化合物c2,所述的制备方法还可进一步包括下述步骤:将如式e所示化合物和叠氮化钠进行如下所示的亲核取代反应,即可;
其中,r3为亲核取代反应中常规的离去基团,例如-ots、-oms、-no2、i-、br-、-otf或-n+me3。
所述亲核取代反应的条件和操作均与本领域该类反应常规的条件和操作相同。
所述制备方法中,当x为18f时,较佳地,还可进一步包括后处理操作。所述后处理的条件和操作与本领域该反应常规的条件和操作相同,较佳地,还可包括以下步骤:进一步分离纯化化合物i,将所述分离纯化后的化合物i富集于sep-pakc18柱,用含有小于10%乙醇的生理盐水淋洗下化合物i,经无菌滤膜过滤。所述分离纯化为本领域常规,较佳地为通过放射性hplc分离纯化,更佳地为先通过sep-pakc18柱纯化或滤膜过滤后再通过放射性hplc分离纯化。
本发明还提供了一种如式i所示的喹唑啉化合物在制备医学显像剂中的应用,其中,x为18f。
如本文所用,f是指自然中稳定存在的19f,自然丰度为100%。
在不违背本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
如无特殊说明,本发明所用试剂和原料均市售可得。
本发明的积极进步效果在于:
1、本发明的pet显像剂制备方法简单,放射性化学产率高,反应时间短,易于工业化大批量生产,更加适合放射药物临床应用。制备得到的18f标记化合物放射化学纯度大于99%。
2、本发明所得到的pet显像剂结构稳定,体内未见脱[18f]氟现象,且显像分辨率高。
3、本发明所得到的pet显像剂具有良好的生物活性,在筛选egfr分子靶向治疗的敏感人群,监测治疗效果以及评估患者预后方面具有巨大的开发潜力。
4、本发明中用到的酪氨酸类化合物前体有商业化供应。
5、本发明中所用到的硫酸铜、抗坏血酸钠等均为商品化试剂,原料廉价易得。
附图说明
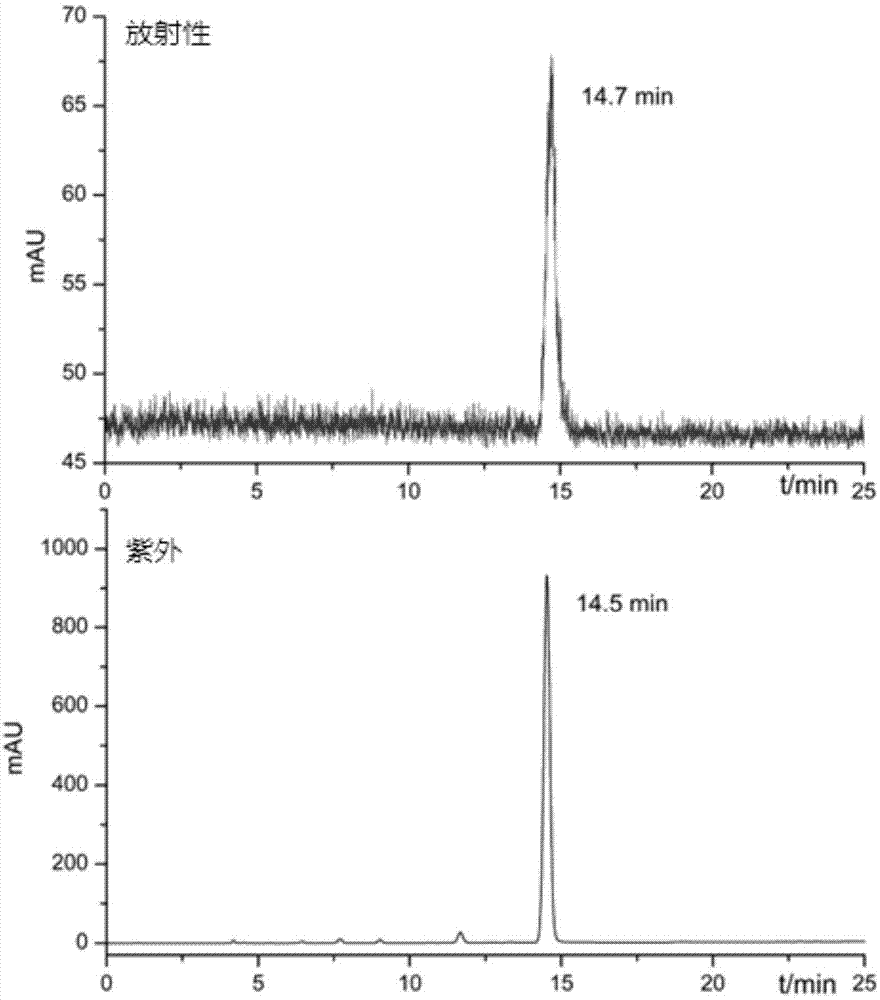
图1为18f化合物i-1与f化合物i-2共同进样的hplc谱图。
图2为实施例2标记产物i-1在pbs中37℃孵育4h时的radio-hplc检测谱图。
图3为实施例2标记产物i-1在pbs中37℃孵育4h时的radio-tlc检测谱图。
图4为实施例2标记产物i-1在小鼠血清中37℃孵育4h时的radio-hplc检测谱图。
图5为实施例2的标记产物i-1在小鼠血清中37℃孵育4h时的radio-tlc检测谱图。
图6为实施例2的标记产物i-1对荷非小细胞肺癌a549肿瘤鼠的pet-ct扫描1.5h时的影像图(横断面)。
图7为实施例2的标记产物i-1对荷非小细胞肺癌a549肿瘤鼠的pet-ct扫描1.5h时的影像图(冠状面)。
图8为实施例2的标记产物i-1在荷非小细胞肺癌a549肿瘤鼠1.5h时的体内分布情况图。
具体实施方式
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
实施例1:i-1的合成
(1)100mci18f-被季铵型阴离子柱qma(美国waters公司产品,18f-由科兴药业有限公司提供)捕获后,取2mlk222(即kryptofix222)溶液(28.8mgk222,6.0mgk2co3,1920μl乙腈,80μl水配成的溶液)将18f冲洗到反应瓶中,反应瓶浸入95℃的油浴,氮气吹干,然后再加入500μl无水乙腈吹干,重复无水乙腈吹干两次;
(2)将含有5mg2-叠氮乙基对甲苯磺酸酯的500μl无水乙腈溶液在氮气保护下迅速加入含有k222、k2co3和18f-的混合物中,95℃下密闭反应10min后停止反应,冰水浴冷却,得到化合物c1-1,标记率可达97%以上;
(3)含有化合物c1-1的反应液在氮气辅助载流下,进行蒸馏,冷凝液收集于含有100μl乙腈的收集瓶中,蒸馏5~20min,得含有提纯的化合物c1-1的乙腈冷凝收集液约600μl,蒸馏效率可达到85%;
(4)向1.5ml指形管中先后加入60μl浓度为0.5mol/l硫酸铜溶液和60μl浓度为1.5mol/l抗坏血酸钠溶液,混匀后加入200μl的pbs溶液(ph=6.0),然后加入100μl含有4.0mg化合物b的dmf溶液,涡旋混合1min后,加入200μl含有活度为5mci的提纯后的化合物c1-1的乙腈溶液,振荡器50℃反应15min;
(5)标记产物用hplc检测并分离(美国agilent1100hplc系统,半制备柱为agilentc18column(9.3mm×250mm)),流动相为水(a)和乙腈(b),梯度分离条件为:0→15→16→23→24min,35%→35%→70%→70%→35%b,流速为2.0ml/min,在进行hplc分离前,反应溶液先用水稀释,用sep-pakc18富集,水洗掉大部分的盐后,用乙腈淋洗下柱子上的富集物,水稀释富集物,然后再hplc进样分离纯化得到化合物i-1,uv(254nm)检测和放射性检测;再将所述分离纯化的产物i-1用水稀释,富集于sep-pakc18柱上,氮气吹干柱子后用乙醇淋洗下产品、溶于生理盐水(使得乙醇含量小于10%),探针浓度为1mci/ml~5mci/ml、经无菌滤膜过滤,得到18f标记酪氨酸激酶类化合物i-1。i-1的标记率>90%,放射化学纯度大于99%,经hplc分离后的放射化学产率~46%(未衰变校正,从18f-计)。
实施例2:i-1的合成
(1)40mci18f-被季铵型阴离子柱qma(美国waters公司产品,18f-由上海科兴药业有限公司提供)捕获后,取1.0mlk222(即kryptofix222)溶液(14.4mgk2.2.2,3.0mgk2co3,960μl乙腈,40μl水配成的溶液)将18f-冲洗到反应瓶中,反应瓶浸入95℃的油浴,氮气吹干,然后再加入500μl无水乙腈吹干,重复上述操作两次;
(2)将含有5mg2-叠氮乙基对甲苯磺酸酯的400μl无水二甲基甲酰胺溶液在氮气保护下迅速加入含有k222、k2co3和18f-的混合物中,100℃下密闭反应5min后停止反应,冰水浴冷却,得到化合物c1-1,标记率可达98%以上;
(3)向含有化合物c-1的反应液中补加乙腈500μl,氮气辅助载流,进行蒸馏,并收集冷凝液,蒸馏5~20min,得含有提纯的化合物c1-1的乙腈冷凝收集液约500μl,蒸馏效率可达到80%;
(4)向1.5ml指形管中先后加入30μl浓度为0.5mol/l硫酸铜溶液和30μl浓度为1.5mol/l抗坏血酸钠溶液,混匀后加入200μl的pbs溶液(ph=6.0),然后加入100μl含有2.0mg化合物b的dmf溶液,涡旋混合1min后,加入250μl含有活度为20mci的提纯后的化合物c1-1的乙腈溶液,振荡器80℃反应10min;
(5)标记产物用hplc检测并分离(美国agilent1100hplc系统,半制备柱为agilentc18column(9.3mm×250mm)),流动相为水(a)和乙腈(b),梯度分离条件为:0→15→16→23→24min,35%→35%→70%→70%→35%b,流速为2.0ml/min,hplc分离前先用滤膜过滤,然后再进样分离纯化后得到化合物i-1,uv(254nm)检测和放射性检测;再将所述分离纯化的产物i-1用水稀释,富集于sep-pakc18柱上,氮气吹干柱子后用乙醇淋洗下产品,溶于生理盐水(使得乙醇含量小于10%),经无菌滤膜过滤,得到18f标记的酪氨酸激酶类化合物,产品最终放射化学产率为71%(衰变校正)。
实施例3:i-2的合成
(1)4.23g2-氟乙基-4甲基苯磺酸酯和3.78g叠氮化钠在dmf中室温反应12h,过滤除去固体后得到c2-1的dmf溶液,产物未进一步纯化,直接用于后面的反应;
(2)在6mlpbs(ph=6)中加入0.5mol/l的硫酸铜和1.5mol/l的抗坏血酸钠各200ul,而后加入26.7mg埃克替尼和2ml上述c2-1的dmf溶液,50℃水浴反应1h。产物加水稀释,乙酸乙酯萃取后干燥,减压浓缩除去乙酸乙酯得到产物i-2,收率为96%。
i-2的鉴定数据如下:
ms(esi):m(c24h25fn6o4)=480.49(m/z),[m+h]+481.15,[m+na]+503.15.
1h-nmr(400mhz,cd3od):δ8.65(s,1h),8.54(s,1h),8.34(s,1h),8.22(s,1h),7.93-7.97(t,1h),7.57-7.58(d,1h),7.46-7.50(t,1h),7.33(s,1h),4.75-4.98(t,2h),4.31-4.34(m,4h),3.77-3.82(m,4h),3.66(m,4h),2.90(s,1h),2.74(s,1h).
13c-nmr(100mhz,cd3od):δ162.79,157.11,156.47,153.82,150.25,146.98,140.50,131.39,130.69,130.11,129.54,122.35,122.07,120.81,119.08,110.74,83.20,81.52,73.42,70.94,70.85,70.43,69.26,68.87.
图1是18f化合物i-1与f化合物i-2共同进样的hplc谱图,时间的偏差是因放射性检测器与紫外检测器串联导致,紫外检测器在前,放射性检测器在后。从图1可以看出,化合物i-1和化合物i-2的保留时间一致,表明两者的结构相同,区别仅在于18f和f。
效果实施例1
实施例1的标记产物i-1在pbs中的稳定性测试:取18f标记制剂50μl(约60μci),置于0.5mlpbs中,置于37℃下,振荡孵育0.5、1、2、3、4h后,用radio-tlc测定其放射化学纯度,以观察其体外稳定性。radio-hplc测定标记产物i-1在pbs中孵育4h后的放射化学纯度。radio-tlc和radio-hplc分析结果显示此实施例的标记化合物未见有放射性杂质产生,表明其在pbs中有良好的稳定性。radio-hplc检测谱图如图2所示,radio-tlc检测谱图如图3所示。
效果实施例2
实施例1的标记产物i-1在血清中的稳定性:取18f标记制剂50μl(约60μci),置于500μl小鼠血清中,置于37℃下,振荡孵育0.5、1、2、3、4h后,用radio-tlc测定其放射化学纯度,以观察其在血清中的稳定性radio-hplc测定标记产物i-1在小鼠血清中孵育4h后的放射化学纯度。hplc分析结果显示此实施例的标记化合物未见有放射性杂质产生,表明其在小鼠血清中有良好的稳定性。radio-hplc检测谱图如图4所示,radio-tlc检测谱图如图5所示。
效果实施例3
实施例1的标记产物i-1的亲脂性(logp)的测定:标记化合物的亲脂性在正辛醇与水溶性缓冲溶液(pbs)组成的体系中测定。该项指标是衡量药物在生物体内的吸收、分布、代谢和消除的一个重要因素。logp为化合物的脂水分配系数,能反映出一个化合物亲脂性的强弱。其测量计算公式为:
logp=log(化合物在正辛醇层的γ计数/化合物在水层的γ计数)
取5μl18f标记制剂于2ml指形管中(含有600μl正辛醇和595μlpbs),密封好,在室温下充分涡旋5min后,高速离心3min,至两相平衡。用移液器从有机相和水相各取样100μl分置于两只γ计数管中,用γ计数器测定计数。根据6次平行实验,计算出logp=1.28。亲脂性数据显示,标记化合物有一定的亲脂性,具有穿过细胞膜进入细胞的能力。
效果实施例4
实施例1的标记产物i-1在荷非小细胞肺癌a549肿瘤鼠的pet/ct显像中的应用:小鼠麻醉后进行ct扫描定位后,把18f标记i-1的制剂200μl(约100μci)通过尾静脉注射到小鼠体内,进行pet扫描成像。整个扫描过程中,保证3l/min的氧气流,混有1.5%的异氟烷。inveonacquisitionworkplace(iaw)控制整个扫描过程。利用osem3d(three-dimensionalorderedsubsetsexpectationmaximum)算法重建图像。pet-ct扫描1.5h时的影像图如图6(横断面)和图7(冠状面)所示。
pet-ct显像表明:pet探针i-1在肿瘤鼠体内主要通过肾脏、肝脏和肠道代谢,体内清除速度较快。a549肿瘤对pet探针i-1有特异性的摄取,肿瘤吸收清晰可见,肿瘤的最大标准摄取值suv为0.65%id/g,平均摄取值suvave为0.35%id/g。i-1在老鼠体内稳定性良好,未发现有脱[18f]氟现象。因此,i-1是一个潜在的靶向egfr的pet探针,可用于表达egfr肿瘤的早期诊断。
效果实施例5
实施例1的标记产物i-1在荷非小细胞肺癌a549肿瘤鼠的体内分布:将200μl约100μci的i-1溶液经尾静脉注射到荷瘤鼠体内,注射1.5h后,通过断颈的方式处死荷瘤鼠并立即解剖。采集感兴趣的脏器或组织,称取样品的质量,并用γ计数器测量其放射性计数。进行衰变校正之后,计算各组织样品的放射性摄取率(%id/g)。体内分布情况如图8所示。
体内分布结果表明,心、脾、肺对探针i-1的摄取均较低,具有较高的靶/非靶比,瘤/肺比值4.3,瘤/心比值4.0,瘤/脾比值6.3。探针主要经肝、肠以及肾清除。该探针对于肺部肿瘤的诊断非常有利。
- 还没有人留言评论。精彩留言会获得点赞!