杂氮硅三环缩水杨醛类锌配合物及其合成方法与流程
本发明涉及一种杂氮硅三环缩水杨醛类锌配合物及其合成方法。
背景技术:
杂氮硅三环类化合物具有免疫增强作用和抗肿瘤作用,对促进细胞增生、伤口愈合有明显作用。杂氮硅三环类化合物如一些席夫碱具有良好的杀菌抗癌作用外,还具有止痛、抗炎、抗菌及灭菌、抗病毒等活性,席夫碱因其结构中的杂化轨道上的n原子具有孤对电子,因此席夫碱结构中可引入各类功能基团使其衍生化,从而在应用上独具特色。水杨醛具有止痛、抗炎、抗菌及灭菌、抗病毒活性,是一种有效的除草剂、杀虫剂、杀菌剂。目前,配合物领域也实现了杂氮硅三环类化合物与水杨醛的合成形成杂氮硅三环类配合物,杂氮硅三环类配合物可用于涂料添加剂,还可起杀菌作用。但是,目前杂氮硅三环类配合物还只是停留在理论研究阶段,并没有一种制备杂氮硅三环类配合物的方法。
技术实现要素:
基于此,有必要提供一种合成成本低、合成时副产物少以及对环境污染小的杂氮硅三环缩水杨醛类锌配合物及其合成方法。
一种杂氮硅三环缩水杨醛类锌配合物的合成方法,包括如下步骤:
取三乙醇胺与γ-氨基丙基三乙氧基硅烷反应,反应产物经过常压蒸馏以蒸出反应产物中的乙醇,将经过常压蒸馏后的残留物再进行减压蒸馏,得到第一中间产物;
取所述第一中间产物,通过无水四氢呋喃溶解得第一溶解液,向所述第一溶解液内滴加水杨醛或者3位取代水杨醛或4位取代水杨醛,室温下搅拌反应,反应后蒸去四氢呋喃,冷却后析出晶体,通过苯与乙醚的混合液对析出的所述晶体进行重结晶得到重结晶体,对所述重结晶体提纯得到第二中间产物;
取醋酸锌并用无水甲醇溶解得第二溶解液,取所述第二中间产物用无水四氢呋喃溶解得第三溶解液,将所述第二溶解液与所述第三溶解液混合均匀后静置反应,析出固体物质,析出的固体物质经过提纯后得到杂氮硅三环缩水杨醛类锌配合物。
在其中一个示例中,3位取代水杨醛或4位取代水杨醛包括:
在其中一个示例中,所述三乙醇胺与γ-氨基丙基三乙氧基硅烷反应时包括如下步骤:
将所述三乙醇胺与γ-氨基丙基三乙氧基硅烷加入第一反应容器中,恒温120℃-140℃下搅拌,反应3h-4h。
在其中一个示例中,所述常压蒸馏以及所述减压蒸馏时包括如下步骤:
对第一反应容器内的反应产物常压蒸馏的温度为90℃-100℃;减压蒸馏时的油浴温度为220℃,收取180℃的馏分,即得所述第一中间产物。
在其中一个示例中,提纯所述重结晶体是:通过用去离子水洗涤多次重结晶体后通过抽滤机抽滤,最后通置入烘箱内干燥得到所述第二中间产物。
在其中一个示例中,所述第二溶解液与所述第三溶解液混合均匀后静置反应时包括如下步骤:
将所述第二溶解液与所述第三溶解液加入第二反应容器中混合均匀并置于阴凉干燥的通风橱内静置反应,直至所述第二反应容器内不再析出所述固体物质。
在其中一个示例中,所述苯与所述乙醚的体积比为1:1-1:2。
在其中一个示例中,所述固体物质通过去离子水洗涤多次后用抽滤机抽干,再置于温度为90℃-100℃的烘箱中烘干,得到所述杂氮硅三环缩水杨醛类锌配合物。
在其中一个示例中,所述醋酸锌与所述第二中间产物反应时的摩尔比为1:2-1:3。
一种所述的杂氮硅三环缩水杨醛类锌配合物的合成方法制得的杂氮硅三环缩水杨醛类锌配合物,所述杂氮硅三环缩水杨醛类锌配合物的化学式如下:
上述杂氮硅三环缩水杨醛类锌配合物的合成方法合成出的杂氮硅三环缩水杨醛类锌配合物的合成率均达到70%以上,合成成本低,合成时副产物少,对环境污染小。合成出的杂氮硅三环缩水杨醛类锌配合物对金黄色葡萄球菌和大肠杆菌有一定的抗菌性能,最低抗菌浓度为1mg/ml。
附图说明
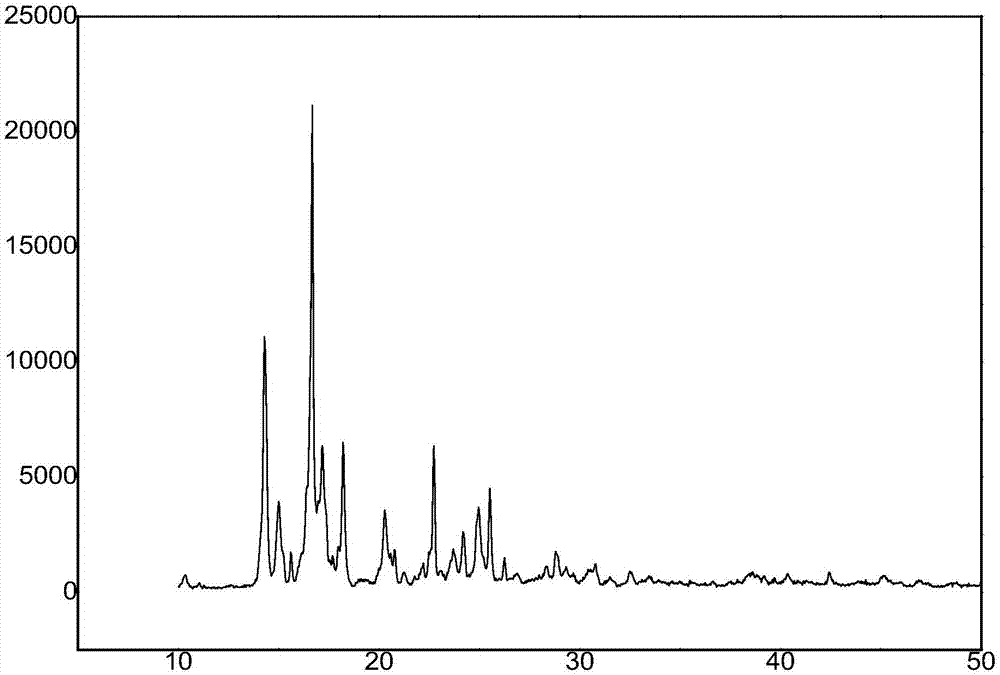
图1为实施例1中的化合物aγ-氨基丙基杂氮硅三环的x射线衍射图;
图2为实施例1中的化合物b3-(2-羟基苯亚甲氨基)丙基杂氮硅三环的x射线衍射图;
图3为实施例1中的化合物c3-(2-羟基苯亚甲氨基)丙基杂氮硅三环锌配合物的x射线衍射图;
图4为实施例1中的化合物b3-(2-羟基苯亚甲氨基)丙基杂氮硅三环的的红外光谱图;
图5为实施例1中的化合物c3-(2-羟基苯亚甲氨基)丙基杂氮硅三环锌配合物的红外光谱图;
图6为实施例1中的化合物b3-(2-羟基苯亚甲氨基)丙基杂氮硅三环的热分析图;
图7为实施例1中的化合物c3-(2-羟基苯亚甲氨基)丙基杂氮硅三环锌配合物的热分析图。
具体实施方式
本发明可以以许多不同的形式来实现,并不限于本文所描述的实施例。相反地,提供这些实施例的目的是使对本发明的公开内容的理解更加透彻全面。
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
本发明提供了一种杂氮硅三环缩水杨醛类锌配合物的合成方法。该杂氮硅三环缩水杨醛类锌配合物的合成方法包括如下步骤:
(1)取三乙醇胺与γ-氨基丙基三乙氧基硅烷反应。三乙醇胺与γ-氨基丙基三乙氧基硅烷反应时包括如下步骤:将三乙醇胺与γ-氨基丙基三乙氧基硅烷加入第一反应容器如圆底烧瓶中,恒温120℃-140℃下搅拌,反应3h-4h。
三乙醇胺与γ-氨基丙基三乙氧基硅烷的反应产物经过常压蒸馏以蒸出反应产物中的乙醇,将经过常压蒸馏后的残留物再进行减压蒸馏,减压蒸馏的油浴温度为220℃,得到第一中间产物。
常压蒸馏以及减压蒸馏时包括如下步骤:在第一反应容器上安装带有干燥管的回流装置,干燥管内填充有氯化钙,常压蒸馏的温度为90℃-100℃;减压蒸馏时的油浴温度为220℃,收取180℃的馏分,即第一中间产物
(2)取第一中间产物,通过无水四氢呋喃溶解得第一溶解液,向第一溶解液内滴加水杨醛或者3位取代水杨醛或4位取代水杨醛,室温下搅拌反应,蒸去四氢呋喃,冷却后析出黄色晶体,通过体积比为1:1-1:2的苯与乙醚的混合液对析出的黄色晶体进行重结晶得到重结晶体,用去离子水洗涤多次如3-5次后通过抽滤机抽滤,最后通置入烘箱内干燥得到第二中间产物(3-(2-羟基苯亚甲氨基)丙基杂氮硅三环、取代水杨醛缩(γ-胺丙基杂氮硅三环)类化合物)。
3位取代水杨醛或4位取代水杨醛包括:
(3)取醋酸锌并用无水甲醇溶解得第二溶解液,取第二中间产物用无水四氢呋喃溶解得第三溶解液,醋酸锌与第二中间产物反应时的摩尔比为1:2-1:3。
将第二溶解液与第三溶解液加入第二反应容器中混合均匀,将第二反应容器塞上带有空隙的塞子后置于阴凉干燥的通风橱内静置反应,析出固体物质直至第二反应容器内不再析出黄色固体物质,析出的黄色固体物质通过去离子水洗涤多次后用抽滤机抽干,再置于温度为90℃-100℃的烘箱中烘干,得到杂氮硅三环缩水杨醛类锌配合物。
杂氮硅三环缩水杨醛类锌配合物的化学式如下:
上述杂氮硅三环缩水杨醛类锌配合物的合成方法合成出的杂氮硅三环缩水杨醛类锌配合物的合成率均达到70%以上,合成率较高,合成时副产物少,对环境污染小。合成出的杂氮硅三环缩水杨醛类锌配合物对金黄色葡萄球菌和大肠杆菌有一定的抗菌性能,最低抗菌浓度为1mg/ml。
以下各实施例中涉及的实验试剂如下表1所示。实验仪器如表2所示。
表1实验试剂
表2实验仪器
实施例1
本实施例提供了一种杂氮硅三环缩水杨醛类锌配合物的合成方法,其包括如下步骤:
(1)取三乙醇胺与γ-氨基丙基三乙氧基硅烷反应。三乙醇胺与γ-氨基丙基三乙氧基硅烷反应时包括如下步骤:将三乙醇胺与γ-氨基丙基三乙氧基硅烷加入第一反应容器中,恒温130℃下搅拌,反应3h。
在第一反应容器上安装带有干燥管的回流装置,干燥管内填充有氯化钙,三乙醇胺与γ-氨基丙基三乙氧基硅烷的反应产物经过常压蒸馏,常压蒸馏的温度为90℃以蒸出反应产物中的乙醇,将经过常压蒸馏后的残留物再进行减压蒸馏,减压蒸馏的油浴温度为220℃,收取180℃的馏分,得到第一中间产物:γ-氨基丙基杂氮硅三环(化合物a)。
反应式为:
(2)取γ-氨基丙基杂氮硅三环,通过无水四氢呋喃溶解得第一溶解液,向第一溶解液内滴加水杨醛,室温下搅拌反应,反应后蒸去四氢呋喃,冷却后析出黄色晶体,通过体积比为1:2的苯:乙醚对析出的黄色晶体进行重结晶得到重结晶体,用去离子水洗涤多次如3-5次后通过抽滤机抽滤,最后通置入烘箱内干燥得到第二中间产物:3-(2-羟基苯亚甲氨基)丙基杂氮硅三环(化合物b)。
化合物b元素分析:anal.calcdforc16h24n2o4si:c57.11,h7.19,n8.32;foundc57.4.18,h7.537,n8.46。1hnmr:8.30(s,1h),3.63(t,2h)1.80(m,2h),0.43(t,2h)。
反应式为:
苯:乙醚体积比为1:2时,对析出的黄色晶体进行重结晶得到重结晶体的重结晶效果好,重结晶体均匀。
苯:乙醚体积比为1:1时,对析出的黄色晶体进行重结晶得到重结晶体的重结晶效果一般。
苯:乙醚体积比为2:1时,对析出的黄色晶体进行重结晶得到重结晶体的重结晶效果较差,重结晶体很难析出。
(3)取醋酸锌并用无水甲醇溶解得第二溶解液,取3-(2-羟基苯亚甲氨基)丙基杂氮硅三环用无水四氢呋喃溶解得第三溶解液,醋酸锌与3-(2-羟基苯亚甲氨基)丙基杂氮硅三环反应时的摩尔比为1:2。
将第二溶解液与第三溶解液加入第二反应容器如锥形瓶中混合均匀,将第二反应容器塞上带有空隙的塞子后置于阴凉干燥的通风橱内静置反应,析出固体物质直至第二反应容器内不再析出黄色的固体物质,析出的黄色的固体物质通过去离子水洗涤多次后用抽滤机抽干,再置于温度为90℃的烘箱中烘干,得到杂氮硅三环缩水杨醛类锌配合物:3-(2-羟基苯亚甲氨基)丙基杂氮硅三环锌配合物(化合物c)。
化合物c元素分析:anal.calcdforc32h46n4o8si2zn:c52.20,h6.30,n7.60;foundc52.48,h6.47,n7.46。1hnmr:8.29(s,1h),3.65(t,2h)1.80(m,2h),0.43(t,2h)。
反应式为:
本发明的种杂氮硅三环缩水杨醛类锌配合物的合成方法的合成副产物较少,产率比较理想,达到70%以上,通过数字显示熔点测试仪分别测得产物的熔点,以及对它们的溶解性能做了一定的测试,数据见表3。
表3化合物性能数据表
实施例2
本实施例提供了一种杂氮硅三环缩水杨醛类锌配合物的合成方法,其包括如下步骤:
(1)取三乙醇胺与γ-氨基丙基三乙氧基硅烷反应。三乙醇胺与γ-氨基丙基三乙氧基硅烷反应时包括如下步骤:将三乙醇胺与γ-氨基丙基三乙氧基硅烷加入第一反应容器中,恒温130℃下搅拌,反应3h。
在第一反应容器上安装带有干燥管的回流装置,干燥管内填充有氯化钙,三乙醇胺与γ-氨基丙基三乙氧基硅烷的反应产物经过常压蒸馏,常压蒸馏的温度为90℃以蒸出反应产物中的乙醇,将经过常压蒸馏后的残留物再进行减压蒸馏,减压蒸馏的油浴温度为220℃,收取180℃的馏分,得到第一中间产物:γ-氨基丙基杂氮硅三环。
(2)取第一中间产物,通过无水四氢呋喃溶解得第一溶解液,向第一溶解液内滴加3位取代水杨醛或者4位取代水杨醛,室温下搅拌反应,蒸去四氢呋喃,冷却后析出晶体,通过体积比为1:2的苯与乙醚的混合液对析出的晶体进行重结晶得到重结晶体,用去离子水洗涤多次如3-5次后通过抽滤机抽滤,最后通置入烘箱内干燥得到第二中间产物:取代水杨醛缩(γ-胺丙基杂氮硅三环)类化合物。
3位取代水杨醛或4位取代水杨醛包括:
(3)取醋酸锌并用无水甲醇溶解得第二溶解液,取取代水杨醛缩(γ-胺丙基杂氮硅三环)类化合物用无水四氢呋喃溶解得第三溶解液,醋酸锌与取代水杨醛缩(γ-胺丙基杂氮硅三环)类化合物反应时的摩尔比为1:2。
将第二溶解液与第三溶解液加入第二反应容器中混合均匀,将第二反应容器塞上带有空隙的塞子后置于阴凉干燥的通风橱内静置反应,析出固体物质直至第二反应容器内不再析出固体物质,析出的固体物质通过去离子水洗涤多次后用抽滤机抽干,再置于温度为90℃的烘箱中烘干,得到取代水杨醛缩(γ-胺丙基杂氮硅三环)锌配合物。
实施例3
本实施例提供了一种杂氮硅三环缩水杨醛类锌配合物,该杂氮硅三环缩水杨醛类锌配合物由实施例2的杂氮硅三环缩水杨醛类锌配合物的合成方法制得。
实施例4
抗菌测试
取实施例2制备出的化合物aγ-氨基丙基杂氮硅三环、化合物b3-(2-羟基苯亚甲氨基)丙基杂氮硅三环以及化合物c3-(2-羟基苯亚甲氨基)丙基杂氮硅三环锌配合物进行抗菌测试。
(1)培养基配制
准确称取3.0g牛肉膏、10.0g蛋白胨、5.0g氯化钠一齐放入烧杯中,量取1000ml水。在上述烧杯中先加入少于所需要的水量,用玻璃棒搅匀,然后加热使其溶解,待完全溶解后补充水到所需体积。然后按1.5%-2.0%的量将琼脂放入已溶的药品中,再加热溶解。加热过程中需要及时补充蒸发掉的水分。溶解完成后,停止加热放置稍微凉一下,但不能让其凝固。
用滴管向培养基中逐滴加入1mol/l氢氧化钠,边加边搅拌,并随时用ph试纸测其ph,直到ph值达到7.4-7.6,然后趁热用滤纸或者多层纱布过滤,将过滤后的液体装入三角烧瓶中(容量不超过1/2)。然后塞上棉塞,并且在棉塞外面包上一层牛皮纸,用麻绳以活结式扎好。
将制好的培养基以0.1mpa,121℃,20min高压蒸汽灭菌。灭菌完成后,取出搁置放凉。再将灭菌后的培养基放入37℃的烘箱中恒温培养24-48h,以检查灭菌是否彻底。如灭菌不彻底需重新配制培养基。
(2)溶液配制
考虑到细菌生物需在有水的条件下才能存活,而化合物b、化合物c无法溶解在水中。因此需要找到既能溶解产物而且还能与水混溶,并且尽可能不具有抗菌性能的溶剂。通过对产物的溶解性可知,化合物b可溶于苯、乙醚、丙酮等有机溶剂,其中丙酮刚好满足实验所需要求。
首先称取1g化合物a溶解在水中,然后转移至100ml的容量瓶中,定容得到10mg/ml的水溶液,记为溶液a。称取1g化合物b将其溶解在大约10ml的丙酮溶液中,由于丙酮既可以溶解产物而且还能与水混溶并且丙酮没有抑菌效应,然后用蒸馏水稀释定容在100ml容量瓶中得到10mg/ml的溶液b。化合物c很难溶解在水和有机溶剂中,只溶解在乙酸溶剂中,但是乙酸本身也有抗菌性能,因此需要一组同浓度下乙酸溶液的对照试验,配制10mg/ml的溶液c以及该溶液下相同浓度的乙酸水溶液,记为溶液d。为了防止产物溶液的水解,需要现配现用。
(3)细菌的选择和纯化
大肠杆菌、金黄色葡萄球菌是实验广泛使用的菌种,其生长繁殖快,易培养,最适生长温度分别为37℃和30℃-37℃。金黄色葡萄球菌可以在15%(2.5mol/l)nacl条件下生长,而大肠杆菌在0.5%-3%nacl条件下生长良好。恰好牛肉膏蛋白胨中的nacl刚好在0.5%,有利于菌种的培养且不干扰实验结果。因此实验选择这两种菌种对其抗菌性能进行测试。
按稀释涂布平板法倒平板,用记号笔标明菌种名称。在近火焰处,左手拿培养皿皿底,右手拿接种环,用接种环按无菌操作挑取菌种,现在培养基的一边作第一次平行划线3次,再转动平板约70°,并将接种环上剩余物质烧掉,待冷却后穿过第一次划线部分进行第二次划线,然后转70°再进行第三次划线和第四次划线,划线完毕后,盖上培养皿盖,倒置于烘箱培养。从分离的平板上单个菌落挑取少许菌苔放入试管进行稀释即得到该菌种的稀释液。
(4)抗菌测试
取八个灭菌的干燥培养皿,分别倒入适量的牛肉膏培养基,待培养基冷却凝固后在四个培养皿上面接种金黄色葡萄球菌(分别记为ⅰ、ⅱ、ⅲ、ⅳ组),剩下的四个培养皿上面接种大肠杆菌(记为ⅴ、ⅵ、ⅶ、ⅷ组)。然后将四组溶液分别稀释成10mg/ml、5mg/ml、1mg/ml和0.5mg/ml四个浓度的试剂。在ⅰ组培养皿上平均取a、b、c、d四个点并用记号笔标注,然后用直径5毫米的单层滤纸片分别沾取10mg/ml的四种试剂即溶液a、溶液b、溶液c、溶液d,同样在ⅱ组培养皿上分别用5mg/ml的四种试剂即溶液a、溶液b、溶液c、溶液d进行测试,
以此类推。将八组培养皿接种完毕后放入37℃烘箱中倒置培养(倒置培养是为了防止水蒸气滴落在培养基上对结果造成干扰)24h。
(5)抗菌实验结果
从实验结果表4和表5可以看出化合物a、化合物b以及化合物c对金黄色葡萄球菌和大肠杆菌都有一定的抗菌性能。
其中抗菌效果比较显著地是产物3-(2-羟基苯亚甲氨基)丙基杂氮硅三环(化合物b),其最低抗菌浓度都大致为0.5mg/ml。产物γ-氨基丙基杂氮硅三环(化合物a)和产物3-(2-羟基苯亚甲氨基)丙基杂氮硅三环锌配合物(化合物c)的抗菌效果差不多,但是抗菌效果没有化合物b显著,最低抗菌浓度均在1mg/ml左右。
根据对照试剂溶液d的抗菌效果来看,大约在5mg/ml以上有一定的抗菌效果,因此当浓度为10mg/ml和5mg/ml时化合物c的抗菌效果均比化合物a的好一些的原因可能是因为醋酸加强了其抗菌性能,起到了协同作用。
由于三种产物均对其有抗菌效果,从而可以得知产物γ-氨基丙基杂氮硅三环、3-(2-羟基苯亚甲氨基)丙基杂氮硅三环和3-(2-羟基苯亚甲氨基)丙基杂氮硅三环锌配合物均对金黄色葡萄球菌和大肠杆菌具有抗菌性能。
表4产物对金黄色葡萄球菌的抗菌效果(直径/毫米)
注:滤纸直径为5毫米
表5产物对大肠杆菌的抗菌效果(直径/毫米)
注:滤纸直径为5毫米
(6)实验分析
1)质谱分析
a、b两个个化合物在电子轰击条件下产生的分子离子及主要碎片离子的m/z和相对丰度数据如表6。根据eims的分子离子和主要碎片离子峰及其相对丰度表可知,杂氮硅三环的电子轰击质谱(eims)表现出一定的特征性,即质谱比较简单,基峰多为m/z174(m-r)+,碎片离子较少且丰度较低。
化合物大部分表现出一定程度的稳定性,存在分子离子峰。分子离子的稳定性与r本身的稳定性和r的电效应有关。r的稳定性或者吸电子诱导效应强都会使得m.+的相对丰度下降。r之所以如此强烈地影响分子离子的稳定性,决定于这类化合物的分子结构。杂氮硅三环中存在着n→si跨环配位键,配位键的强弱势必将影响分子的稳定性,因此质谱的分子离子丰度也有所反映,n→si建的强弱可以键长表示。n→si键的强度大,则键长短;反之则长。
n→si配位键长短决定于取代基r的电效应(在不存在空间效应的情况下)。如r为吸电子基团,由于r的诱导效应,使得硅原子上的电子密度移向r,使其正电性增加、接受电子能力变大,形成的n→si配位键较强,键长也就短。结果是在电子轰击条件下,更为倾向发生si-r键的断裂,生成具有n-si共价键的稳定杂氮硅三环离子[基峰为m/z174(m-r)+],也就相对地削弱了m.+的丰度。
表6eims的分子离子和主要碎片离子峰及其相对丰度
2)x射线衍射分析
由化合物a-化合物c的x射线衍射分析图谱(参见图1-图3所示)分别得到三组三强峰,然后根据公式2dsinθ=nλ(n为任意正整数,称为该晶面的一级、二级…n级反射,为了方便将其改写为2dhklsinθ=λ,其中dhkl=dhkl/n并且n=1的一级反射)。求得图1中三强峰的d值分别为
由于硅的最外层具有3d空轨道,轨道杂化形式之一是sp3d,其中si←n配位键是由于硅原子上的3d空轨道接受氮原子的p孤对电子所形成的,这就是产生五配位硅的化合物的原因。从而形成了特殊的si←n配位键。配位数为5的硅原子以sp3d杂化轨道成键,分子成三角双锥构型,三个氧原子和硅处于同一平面,彼此之间夹角为120°,氮原子和r基团分别处于平面上下键的两个项端位置,硅原子接受氮上的孤对电子形成跨环si←n配位键。以si←n键为轴组成三个五元环的笼状对称结构。
3)红外光谱分析
结合图4和图5所示,通过查找红外光谱基团的吸收频率可以看出,产物均在580cm-1-590cm-1之间有两组中等强度的吸收峰,这是杂氮硅特有的红外吸收峰,因为杂氮硅中的si←n配位键在该强度下有吸收峰,而普通的si-n键的吸收频在780cm-1-800cm-1,说明产物都存在硅氮配位键。
在750cm-1和1100cm-1附近有两组较强的吸收峰,这是si-o键的特征吸收峰;在1260cm-1处附近有si-c键的特征吸收峰。
1640cm-1处出现了ch=n的特征吸收峰。图4和图5均可以看出在3030cm-1-3070cm-1区域产生一组吸收带,这是苯环上的c-h伸缩振动和苯环骨架振动的和频,图4和图5在3500cm-1虽然均有吸收峰,但是图4其吸收峰比较宽大而图5处的吸收峰显然稍显尖锐,这是由于图4中3500cm-1处的吸收峰是-oh基的特有吸收带,而图5中3500cm-1处的吸收峰是因为羟基中的氢原子被锌取缔。
5)化合物的热分析
参见图6及图7所示,对化合物b和化合物c做了热分析(dsc和tg),从配体和配合物的热分析图可以明显看出,二者的热重曲线明显不同。配体前期失重较快,而配合物在前期失重速度要慢些。说明形成配合物以后热稳定性提高。
(7)结论
a、通过该实验合成了γ-氨基丙基杂氮硅三环、3-(2-羟基苯亚甲氨基)丙基杂氮硅三环和3-(2-羟基苯亚甲氨基)丙基杂氮硅三环锌配合物三个产物,产物合成率均达到70%以上,合成率较高,且合成时副产物较少,对环境友好。
b、γ-氨基丙基杂氮硅三环可溶解在水中,以及大部分有机溶剂,溶解性能较好。而3-(2-羟基苯亚甲氨基)丙基杂氮硅三环和3-(2-羟基苯亚甲氨基)丙基杂氮硅三环锌配合物不溶于水,前者可溶于苯、乙醚、丙酮等有机溶剂,而后者仅仅溶于乙酸溶剂。
c、三者均对金黄色葡萄球菌和大肠杆菌有一定的抗菌性能。其中抗菌性能较显著的是3-(2-羟基苯亚甲氨基)丙基杂氮硅三环,最低抗菌浓度为0.5mg/ml。γ-氨基丙基杂氮硅三环和3-(2-羟基苯亚甲氨基)丙基杂氮硅三环锌配合物抗菌性能相仿,最低抗菌浓度为1mg/ml。
d、通过对其进行质谱、x射线衍射和红外检测可发现产物都存在特殊的si←n配位键,从而具有相似的性能,但由于r基团的不同,又各自有着特殊的性能。
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!