一种吡唑并-吡啶酮化合物晶型D的制作方法
本发明属于晶型药物分子技术领域,特别涉及一种吡唑并-吡啶酮化合物晶型d。
背景技术:
阿哌沙班(apixaban商品名eliquis,),其化学名称为1-(4-甲氧基苯基)-7-氧代-6-[4-(2-氧代-1-哌啶基)苯基]-4,5,6,7-四氢-1h-吡唑并[3,4-c]吡啶-3-甲酰胺,cas:503612-47-3,其结构如式i所示。阿哌沙班是辉瑞与百时美施贵宝联合开发的口服的选择性活化xa因子抑制剂,可直接抑制凝血因子xa,阻断凝血级联过程中凝血酶原转化成凝血酶,而且阿哌沙班对xa选择性高、作用强。除此之外,阿哌沙班不仅可抑制游离的xa及凝血酶原复合物中的xa,还可抑制血凝块中的xa,且在抑制过程中无需抗凝血酶ⅲ,这与肝素类抗凝药如磺达肝癸钠的作用不同。2011年5月,阿哌沙班被批准在欧盟上市,用于预防接受择期髋关节或膝关节置换术的成年患者出现静脉血栓栓塞症(vte)事件;2012年12月美国fda批准该药物用于降低非瓣膜房颤患者脑卒中和全身性栓塞风险。化学结构式如下所示:
同一药物的不同晶型在溶解度、熔点、密度、热稳定性,化学反应性、光学和机械性质等方面有不同的特性,这些特性可以直接影响药物的稳定性、均一性、生物利用度、疗效和安全性。因此,药物研发中进行全面系统的多晶型筛选,选择最适合开发的晶型,是不可忽视的重要研究内容之一。
us20060160841首先公开了阿哌沙班的非溶剂晶型n-1和二水合物晶型h2-2,并在专利文献wo2007001385及其cn101065379a公开了晶型n-1和晶型h2-2的具体晶胞参数、位置坐标参数、x-射线衍射特征峰位置、ssnmr(固体核磁共振)位移等晶体表征参数。
us20070203178公开了阿哌沙班的n,n-二甲基甲酰胺溶剂合物晶型dmf-5和甲酰胺溶剂合物晶型fa-2。
wo2012168364则详细公开了阿哌沙班的晶型α及其表征。
专利cn102770126和专利cn103830199公开了阿哌沙班的多种晶型,但是这些发明工艺生产效率低,产品收率低,不适合工业化生产。
目前公开的阿哌沙班晶型溶解性差,影响药物的疗效和临床效果,因此,需要开发更多的晶型,一方面为药物应用提供更多的阿哌沙班晶型,另一方面,也要开发更适合工业化生产的,经济效益高的阿哌沙班晶型。
技术实现要素:
鉴于现有技术的不足,本申请一方面提供一种吡唑并-吡啶酮化合物晶型d。
本申请中所指的吡唑并-吡啶酮化合物晶型d是阿哌沙班晶型d,所指的吡唑并-吡啶酮化合物是阿哌沙班。
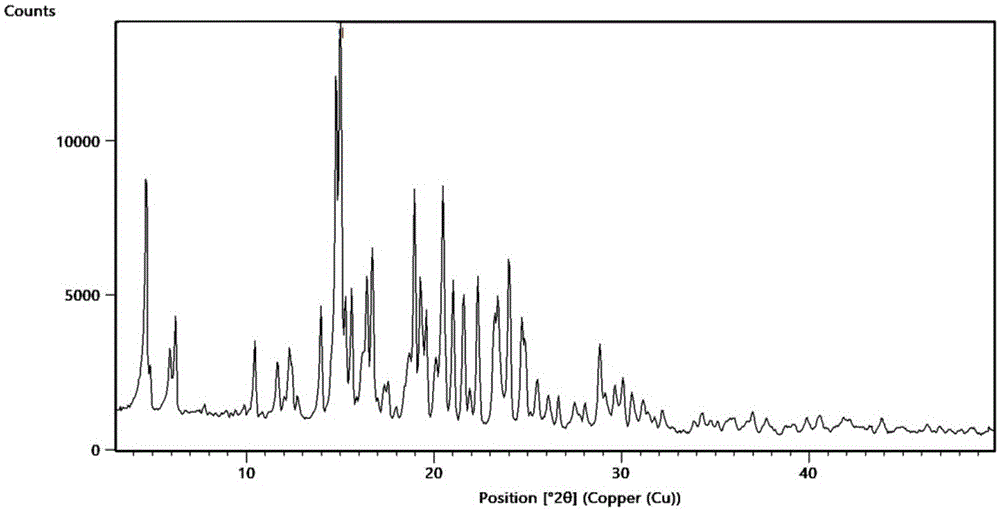
根据本发明的第一方面,提供了一种吡唑并-吡啶酮化合物晶型d。所述的吡唑并-吡啶酮化合物晶型,使用cu-kα辐射,以2θ表示的x射线衍射谱图在5.8±0.2°,15.1±0.2°,16.2±0.2°,20.2±0.2°,21.7±0.2°,22.8±0.2°,24.6±0.2°,25.2±0.2°处有特征峰。
优选地,所述的吡唑并-吡啶酮化合物晶型d,使用cu-kα辐射,以2θ表示的x射线衍射谱图在5.8±0.2°,7.1±0.2°,7.4±0.2°,11.6±0.2°,12.8±0.2°,15.1±0.2°,16.2±0.2°,18.5±0.2°,20.2±0.2°,21.7±0.2°,22.8±0.2°,23.5±0.2°,24.6±0.2°,25.2±0.2°处有特征峰。
优选地,所述的吡唑并-吡啶酮化合物晶型d,使用cu-kα辐射,以2θ表示的x射线衍射谱图在5.8±0.2°,7.1±0.2°,7.4±0.2°,11.6±0.2°,12.8±0.2°,15.1±0.2°,16.2±0.2°,16.8±0.2°,17.6±0.2°,17.9±0.2°,18.5±0.2°,20.2±0.2°,20.5±0.2°,21.7±0.2°,22.2±0.2°,22.8±0.2°,23.5±0.2°,24.4±0.2°,24.6±0.2°,25.2±0.2°,26.1±0.2°,30.1±0.2°,31.3±0.2°处有特征峰
优选地,所述的吡唑并-吡啶酮化合物晶型d,使用cu-kα辐射,其特征峰符合如图1所示的x射线粉末衍射图谱。
优选地,所述的吡唑并-吡啶酮化合物晶型d,其差示扫描量热分析检测在176±2℃出现放热峰,且在239±2℃出现吸热峰。
本发明的第二方面提供一种吡唑并-吡啶酮化合物晶型d的制备方法,具体制备步骤包括:
(1)将吡唑并-吡啶酮化合物加入到n-甲基吡咯烷酮溶液中,加热使其完全溶解;
(2)将步骤(1)中溶液降温后加入有机溶剂a和水的混合溶液,保温搅拌;
(3)将步骤(2)中溶液降温析晶,过滤,干燥,得吡唑并-吡啶酮化合物晶型d。
所述的制备方法,步骤(1)中吡唑并-吡啶酮化合物与n-甲基吡咯烷酮的质量比为1:5~20。
所述的制备方法,步骤(2)中有机溶剂a的用量为吡唑并-吡啶酮化合物质量的2.5~45倍。
所述的制备方法,步骤(2)中吡唑并-吡啶酮化合物与水的质量比为1:0.1~5。
所述的制备方法,步骤(2)中有机溶剂a选自乙二醇,丙二醇,苯甲醇,异丙醚,乙二醇二甲醚,丙酮,甲酰胺,乙腈,四氢呋喃,二氯甲烷,乙酸中的一种或多种。
优选地,步骤(2)中有机溶剂a选自苯甲醇,异丙醚,乙二醇二甲醚,丙酮,乙腈,二氯甲烷中的一种或多种。
所述的制备方法,步骤(2)中降温温度为30~45℃。
所述的制备方法,步骤(3)中降温析晶的温度为-15~10℃。
以下内容进一步详述吡唑并-吡啶酮化合物晶型d的制备方法:
(1)将吡唑并-吡啶酮化合物加入到n-甲基吡咯烷酮溶液中,加热使其完全溶解;
(2)将步骤(1)中溶液降温至30~45℃后加入有机溶剂a和水的混合溶液中,保温搅拌2~6h;
(3)将步骤(2)中溶液降温至-15~10℃,析晶1~5h,过滤,干燥,得吡唑并-吡啶酮化合物晶型d。
优选地,有机溶剂a的用量为吡唑并-吡啶酮化合物质量的10~30倍。
优选地,步骤(3)中降温析晶的温度为-8~6℃。
进一步优选地,步骤(3)中降温析晶的温度为-2~3℃。
本发明的第三方面提供一种药物组合物,该组合物包含本发明所述的吡唑并-吡啶酮化合物晶型d,并含有其它药学上可接受的辅料组分。
优选地,本发明的药物组合物制备如下:使用标准和常规的技术,使本发明化合物与制剂学上可接受的固体或液体载体结合,以及使之任意地与制剂学上可接受的辅助剂和赋形剂结合制备成可用剂型。
优选的,所述的其它组分包括可联合使用的其它活性成分、赋形剂、填充剂等。
优选的,所述的药物组合物为喷雾剂、片剂、胶囊剂、粉针剂、液体注射剂等。
本发明中x-射线粉末衍射测试仪器及测试条件:x-射线粉末衍射仪:panalyticale;cu-kα;样品台:平板;入射光路:bbhd;衍射光路:plxcel;电压45kv,电流40ma;发散狭缝:1/4;防散射狭缝:1;索拉狭缝:0.04rad;步长:0.5s;扫描范围:3~50°。其对应的x射线粉末衍射图(cu-kα)中特征峰详见附图1及表1。
表1吡唑并-吡啶酮化合物晶型d的pxrd峰
本发明中dsc热分析测试仪及测试条件:tga/dsc热分析仪:mettlertoledotga/dsc3+;动态温度段:30~350℃;加热速率:10℃/min;程序段气体n2;气体流量:50ml/min;坩埚:铝坩埚40μl。
本发明所述方法制备的吡唑并-吡啶酮化合物晶型的tga/dsc测试结果如图2所示。dsc结果表明本发明制备的吡唑并-吡啶酮化合物晶型不含溶剂和水。
结合tga/dsc检测结果以及x射线粉末衍射结果表明,本发明制备的晶型为吡唑并-吡啶酮化合物新晶型。
本发明所述方法制备的吡唑并-吡啶酮化合物晶型d相对于目前报道的吡唑并-吡啶酮化合物晶型具有以下优势:
(1)收率高、纯度高。收率大于89%,纯度高于99.89%。
(2)溶解性好。本发明制备的吡唑并-吡啶酮化合物晶型d相对于现有晶型,在不同溶解介质中均具有较高的溶解度,是现有吡唑并-吡啶酮化合物晶型溶解度的1.5倍以上。
(3)溶出度高。使用本发明制备的吡唑并-吡啶酮化合物晶型d做成的片剂,相对于现有晶型做成的片剂,吡唑并-吡啶酮化合物有效成分溶出快、溶出比例高。
(4)本发明制备方法简便,对设备要求较低,成本低,适合于高纯度,高产率的吡唑并-吡啶酮化合物工业化生产,具有很高的经济效益。
附图说明
图1:吡唑并-吡啶酮化合物晶型d的x射线粉末衍射图谱。
图2:吡唑并-吡啶酮化合物晶型d的差示扫描量热曲线(dsc)图。
具体实施方式
现通过以下实施例来进一步描述本发明的有益效果,实施例仅用于例证的目的,不限制本发明的范围,同时本领域普通技术人员根据本发明所做的显而易见的改变和修饰也包含在本发明范围之内,吡唑并-吡啶酮化合物为阿哌沙班。
实施例1:
(1)将10.0g吡唑并-吡啶酮化合物加入到120gn-甲基吡咯烷酮溶液中,加热到100℃使其完全溶解;
(2)将步骤(1)中溶液缓慢降温至45℃后,加入110g混合溶剂(100g丙酮+10g纯化水),保温搅拌4h;
(3)将步骤(2)中溶液降温至0℃,搅拌析晶5h,过滤,收集析出的结晶体,55℃真空干燥,即得9.6g吡唑并-吡啶酮化合物晶型d化合物,收率96.0%,hplc纯度99.98%。
实施例2:
(1)将10.0g吡唑并-吡啶酮化合物加入到50gn-甲基吡咯烷酮溶液中,加热到110℃使其完全溶解;
(2)将步骤(1)中溶液缓慢降温至40℃后,加入210g混合溶剂(180g乙腈+30g纯化水),保温搅拌2h;
(3)将步骤(2)中溶液降温至3℃,搅拌析晶4h,过滤,收集析出的结晶体,55℃真空干燥,即得9.57g吡唑并-吡啶酮化合物晶型d化合物,收率95.7%,hplc纯度99.96%。
实施例3:
(1)将10.0g吡唑并-吡啶酮化合物加入到200gn-甲基吡咯烷酮溶液中,加热到95℃使其完全溶解;
(2)将步骤(1)中溶液缓慢降温至30℃后,加入305g混合溶剂(300g乙二醇二甲醚+5g纯化水),保温搅拌6h;
(3)将步骤(2)中溶液降温至-2℃,搅拌析晶1h,过滤,收集析出的结晶体,55℃真空干燥,即得9.45g吡唑并-吡啶酮化合物晶型d化合物,收率94.5%,hplc纯度99.95%。
实施例4:
(1)将10.0g吡唑并-吡啶酮化合物加入到150gn-甲基吡咯烷酮溶液中,加热到120℃使其完全溶解;
(2)将步骤(1)中溶液缓慢降温至45℃后,加入26g混合溶剂(25g异丙醚+1g纯化水),保温搅拌5h;
(3)将步骤(2)中溶液降温至6℃,搅拌析晶1h,过滤,收集析出的结晶体,55℃真空干燥,即得9.41g吡唑并-吡啶酮化合物晶型d化合物,收率94.1%,hplc纯度99.94%。
实施例5:
(1)将10.0g吡唑并-吡啶酮化合物加入到90gn-甲基吡咯烷酮溶液中,加热到85℃使其完全溶解;
(2)将步骤(1)中溶液缓慢降温至40℃后,加入500g混合溶剂(450g苯甲醇+50g纯化水),保温搅拌3h;
(3)将步骤(2)中溶液降温至-8℃,搅拌析晶2h,过滤,收集析出的结晶体,55℃真空干燥,即得9.35g吡唑并-吡啶酮化合物晶型d化合物,收率93.5%,hplc纯度99.93%。
实施例6:
(1)将10.0g吡唑并-吡啶酮化合物加入到70gn-甲基吡咯烷酮溶液中,加热到70℃使其完全溶解;
(2)将步骤(1)中溶液缓慢降温至30℃后,加入180g混合溶剂(140g丙酮+40g纯化水),保温搅拌5h;
(3)将步骤(2)中溶液降温至-15℃,搅拌析晶4h,过滤,收集析出的结晶体,55℃真空干燥,即得9.26g吡唑并-吡啶酮化合物晶型d化合物,收率92.6%,hplc纯度99.92%。
实施例7:
(1)将10.0g吡唑并-吡啶酮化合物加入到250gn-甲基吡咯烷酮溶液中,加热到90℃使其完全溶解;
(2)将步骤(1)中溶液缓慢降温至20℃后,加入180g混合溶剂(90g苯甲醇+90g二氯甲烷),保温搅拌8h;
(3)将步骤(2)中溶液降温至-5℃,搅拌析晶5h,过滤,收集析出的结晶体,55℃真空干燥,即得9.14g吡唑并-吡啶酮化合物晶型d化合物,收率91.4%,hplc纯度99.91%。
实施例8:
(1)将10.0g吡唑并-吡啶酮化合物加入到80gn-甲基吡咯烷酮溶液中,加热到130℃使其完全溶解;
(2)将步骤(1)中溶液缓慢降温至60℃后,加入860g混合溶剂(800g乙二醇+60g纯化水),保温搅拌1h;
(3)将步骤(2)中溶液降温至10℃,搅拌析晶0.5h,过滤,收集析出的结晶体,55℃真空干燥,即得9.04g吡唑并-吡啶酮化合物晶型d化合物,收率90.40%,hplc纯度99.90%。
实施例9:
(1)将10.0g吡唑并-吡啶酮化合物加入到100gn-甲基吡咯烷酮溶液中,加热到130℃使其完全溶解;
(2)将步骤(1)中溶液缓慢降温至50℃后,加入420g混合溶剂(300g丙二醇+120g纯化水),保温搅拌3h;
(3)将步骤(2)中溶液降温至-10℃,搅拌析晶0.5h,过滤,收集析出的结晶体,55℃真空干燥,即得8.9g吡唑并-吡啶酮化合物晶型d化合物,收率89.0%,hplc纯度99.89%。
实施例10:
(1)将10.0g吡唑并-吡啶酮化合物加入到40gn-甲基吡咯烷酮溶液中,加热到130℃使其完全溶解;
(2)将步骤(1)中溶液缓慢降温至70℃后,加入500g混合溶剂(400g四氢呋喃+100g纯化水),保温搅拌2h;
(3)将步骤(2)中溶液降温至5℃,搅拌析晶1.5h,过滤,收集析出的结晶体,55℃真空干燥,即得9.06g吡唑并-吡啶酮化合物晶型d化合物,收率90.6%,hplc纯度99.90%。
实施例11:
(1)将10.0g吡唑并-吡啶酮化合物加入到250gn-甲基吡咯烷酮溶液中,加热到110℃使其完全溶解;
(2)将步骤(1)中溶液缓慢降温至70℃后,加入350g混合溶剂(200g甲酰胺+150g纯化水),保温搅拌2h;
(3)将步骤(2)中溶液降温至5℃,搅拌析晶1.5h,过滤,收集析出的结晶体,55℃真空干燥,即得8.91g吡唑并-吡啶酮化合物晶型d化合物,收率89.1%,hplc纯度99.85%。
实施例12:
(1)将10.0g吡唑并-吡啶酮化合物加入到150gn-甲基吡咯烷酮溶液中,加热到120℃使其完全溶解;
(2)将步骤(1)中溶液缓慢降温至70℃后,加入300g混合溶剂(100g四氢呋喃+200g纯化水),保温搅拌2h;
(3)将步骤(2)中溶液降温至10℃,搅拌析晶1.5h,过滤,收集析出的结晶体,55℃真空干燥,即得8.93g吡唑并-吡啶酮化合物晶型d化合物,收率89.3%,hplc纯度99.87%。
对比实施例1:
室温下,加入阿哌沙班10g和n,n-二甲基甲酰胺50g,搅拌,升温到80~90℃溶解澄清,加入1%活性炭脱色30分钟,热过滤得到澄清溶液;将以上得到的阿哌沙班澄清溶液保温在80℃滴加异丙醇20g,转速30转/分,搅拌养晶20分钟,继续保温滴加80g异丙醇,滴加90min完毕,搅拌1小时,自然降温到室温,过滤,用10g异丙醇洗涤固体,固体在70℃真空干燥得到阿哌沙班白色带光泽固体,收率80.1%,hplc纯度90.0%。
对比实施例2:
取阿哌沙班3.5g于适当体积的圆底烧瓶中,加入1000ml乙腈,回流温度下搅拌溶解,
过滤除去杂质,将滤液放入冷冻干燥机中,-80℃冷冻干燥,得3.0g阿哌沙班晶体ⅰ,产率85.7%,hplc纯度97.2%。
对比实施例3:
取阿哌沙班2g于适当体积的圆底烧瓶中,加入500ml乙腈,回流温度下搅拌溶解,过滤除去杂质,将滤液利用旋转蒸发仪快速除去溶剂,室温干燥,得1.72g阿哌沙班晶体ⅱ,产率86.2%,hplc纯度97.7%。
对比实施例4:
在500ml反应瓶中投入非b晶型阿哌沙班(2g),加入四氢呋喃(350ml),搅拌加热至固
体全部溶解,降温至40℃以下,加入阿哌沙班b晶型晶种,缓慢冷却至室温,加入100ml水,
继续冷却至0℃析晶,抽滤,烘干得到1.63g阿哌沙班b晶型产品,收率81.5%,hplc纯度:95.6%。
对比实施例5:
将5.0g阿哌沙班粗品加入到50ml乙腈和50ml乙醇混合溶剂中,加热至溶解,降温至0~5℃析晶2h,抽滤,50℃真空干燥,得到4.0g白色γ晶型的阿哌沙班,收率约80.1%,hlpc纯度97.4%。
对比实施例6:
将1.00g阿哌沙班粗品加至反应瓶中,加入8.0ml的1,3-丙二醇,搅拌加热至120℃,全溶后继续加热搅拌0.5h,停止加热自然降温析出晶体。当反应瓶内温度为55℃时,加入10ml水,继续搅拌,降至室温时,冰水浴降温至10℃,搅拌析晶2h。抽滤,水洗(3×5ml),65℃真空干燥30h,得阿哌沙班γ晶型产品0.86g,收率为86.0%,hplc纯度为96.8%。
对比实施例7:
将17g阿哌沙班粗品加入到装有120ml乙酸乙酯的三口烧瓶中,加热至70℃,搅拌2小时,将悬浮液降温冷却至40℃后,用高速剪切机10000转/min的转速下对其剪切10min后。过滤,所得滤饼在50℃下真空干燥3小时,得到12.3g白色阿哌沙班晶型i,收率72.4%。hplc纯度:95.1%。
对比实施例8:
将纯度为98.2%的阿哌沙班粗品4.35g中加入130ml质量分数为6%的乙酸的甲醇溶液,
加热至回流溶清,而后搅拌自然降温至0~5℃析晶,过滤,真空干燥后得3.4g类白色阿哌沙班晶体。结晶收率:78.1%,hplc纯度:98.8%。
对比实施例9:
在湿度大于75%的环境下,将阿哌沙班加入冰醋酸中,加热溶解或者搅拌悬浮;缓慢冷却至室温,析出固体;过滤,洗涤,40℃真空干燥,真空干燥至两次称重(间隔1小时左右)差值小于0.5%为恒重,得产物阿哌沙班一水合物,收率81.6%,hplc纯度:93.7%。
对比实施例10:
将阿哌沙班(0.5g)加入到乙酸(2ml)中,加热至80℃,搅拌形成溶液。将乙酸乙酯(10ml)缓慢加入到溶液中,待大量固体析出,慢慢降温至30℃,并搅拌3小时。过滤,收集的固体在80℃真空干燥,得到阿哌沙班(0.35g),收率为70%,hplc纯度为97.8%。
溶解性实验
具体的溶解度试验参考中国药典2015。分别精密称取实施例1~12和对比实施例1~10的阿哌沙班过量,置于小西林瓶中,分别加入水、0.1mol/l盐酸、ph6.8的磷酸盐缓冲溶液,配置成阿哌沙班饱和溶液,摇匀溶解,过滤,照紫外-可见分光光度法(通则0401),在280nm的波长处测定吸光度来计算其溶解度,结果见表2。
表2不同阿哌沙班晶型在不同介质中的溶解度
经试验,本发明方案制备的所有阿哌沙班晶型d均具有相近的溶解度效果。由表2可见,本发明方案制备的阿哌沙班在不同ph溶液中的溶解度均高于对比例1至对比例10晶型的溶解度,本发明制备的阿哌沙班晶型相对于现有晶型,具有较高的溶解性。
溶出度试验
分别使用实施例1~12和对比实施例1~10制得的阿哌沙班晶体,以干法制粒的方法,按照表3的配方比例分别制备1000片阿哌沙班(规格为5mg/片)片剂。
表3含阿哌沙班晶型的药物制剂配比
参考fda的溶出度测试方法及取样时间,使用usp装置2(桨)方法以75rpm的旋转速度在0.1mol/l盐酸溶出介质中在37℃进行溶出测试。测试开始10、20、30、45和60分钟后移去样品并通过hplc在波长280nm处分析阿哌沙班。结果见表4。
表4不同阿哌沙班晶型制备的片剂在0.1mol/l盐酸中的溶出度
经试验,本发明方案制得的所有阿哌沙班晶型d制备的片剂均具有相近的溶出度效果。由表4可见,用本发明技术制备的阿哌沙班晶型做成的片剂,相对比于现有的阿哌沙班晶型做成的片剂,在0.1mol/l盐酸溶液中,具有较高的溶出度。
- 还没有人留言评论。精彩留言会获得点赞!