一种异辛酸铜的制备方法与流程
本发明涉及一种有机酸金属盐的制备方法,具体涉及一种异辛酸铜的制备方法。
背景技术:
异辛酸铜在传统行业中用于:用于船底漆防污剂、海底电缆和一般电缆的防水剂;不饱和聚酯的抑制剂;其它制品的防霉、防腐剂等。但是,近年来异辛酸铜在有机金属催化行业能解决一些其他方案无法克服的偶联应用,尤其是在氧化偶联方面。
异辛酸铜作为催化剂可以提高有机合成的效率,直接构建c-c键和c-n键,缩短反应步骤。但是,对于异辛酸铜的制备方法,在传统文献中并没有异辛酸铜的合成方案。
技术实现要素:
本发明提供一种异辛酸铜的制备方法,该方法解决了现在没有异辛酸铜的合成方案问题,能够通过打浆得到纯度较高的异辛酸铜,该方法对设备要求不高,原料成本低廉,操作简单,条件易控,且所得产品催化效率高,绿色高效。
为了达到上述目的,本发明提供了一种异辛酸铜的制备方法,该方法包含:合成异辛酸负离子,通过异辛酸负离子和铜源在50~100℃下于水和与水不混溶的有机溶剂a中反应;待反应结束,将有机相去除大部分溶剂,加入有机溶剂b在20℃~40℃进行打浆或重结晶,过滤,干燥,得到固体异辛酸铜。其中,在进行打浆或重结晶时,较低温度,体系容易成胶体状物质,较高温度产品溶解性较多,故在20℃~40℃进行打浆或重结晶。
其中,所述的有机溶剂a包含:正己烷、正庚烷、沸程为60~90℃的石油醚、甲苯和二甲苯中的任意一中或两种以上;所述有机溶剂b包含:甲醇、乙腈、丙酮和正己烷中的任意一中或两种以上。选择溶剂时,不能选择乙酸乙酯、dmf、甲苯和thf等溶剂,不适用于结晶或者打浆析出。
优选地,所述异辛酸负离子的合成方法为:将异辛酸和水搅拌混合,滴加入无机碱的水溶液,在50~100℃下反应,得到异辛酸负离子。
优选地,所述无机碱包含:naoh、koh、na2co3、k2co3、ca(oh)2、nah和cah中的任意一种或两种以上。
优选地,所述异辛酸和naoh的摩尔比为1.1:1,所述naoh与水的质量比为1:10。
优选地,所述异辛酸和无机碱的水溶液在75~100℃下反应2~5h。
优选地,所述铜源包含:硫酸铜、氯化铜、溴化铜、碘化铜或其水合物中任意一中或两种以上。
优选地,所述异辛酸负离子和铜源在50~100℃下于水和与水不混溶的有机溶剂a中反应3~10h。
优选地,所述异辛酸负离子和铜源在75~100℃下于水和与水不混溶的有机溶剂a中反应3~10h。
优选地,所述异辛酸负离子和铜源反应结束后,分液,将水相用有机溶剂a萃取,合并有机相,将有机相用40~50℃的热水萃取洗涤,然后去除大部分溶剂。
优选地,所述异辛酸和铜源的摩尔比为2.2:1。
本发明提供的异辛酸铜的制备方法,解决了现在没有异辛酸铜的合成方案问题,具有以下优点:
本发明的方法通过打浆或者重结晶得到纯度较高的异辛酸铜,该方法对设备要求不高,原料成本低廉,操作简单,条件易控,且所得产品催化效率高,绿色高效。
附图说明
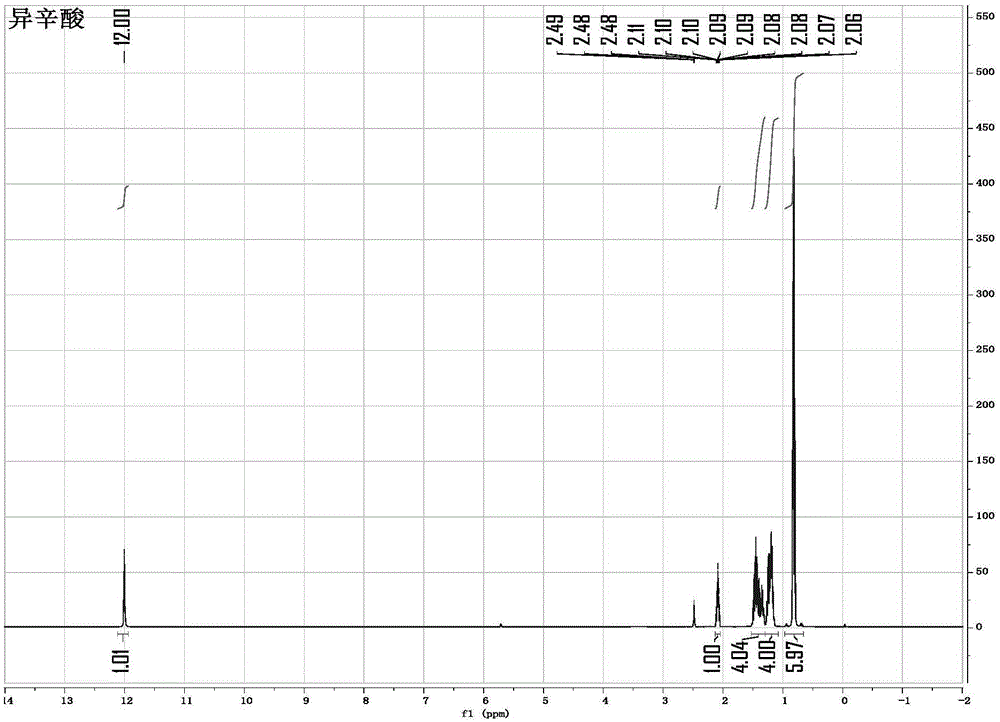
图1为异辛酸的1hnmr数据谱图。
图2为本发明实施例1制备的异辛酸铜的1hnmr数据谱图。
具体实施方式
下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
取88.8g异辛酸(0.62mol,2.20equiv)和88.8g水,加入反应器(或反应釜)中,搅拌体系。称取22.4gnaoh(0.56mol,2.00equiv),溶于224.0g水中,逐渐滴加至反应器中,然后对体系升温至75℃,并维持此温度2.5h。
取800.0ml甲苯加入到反应体系中。称取44.8gcuso4(0.28mol,1.00equiv)溶于267.0g水中,逐渐滴加至反应器中,滴加完毕后,搅拌反应7.5h。
冷却上述反应体系至室温,分液,分出有机相,水相用300ml/次的甲苯萃取两次,合并有机相。有机相用40~50℃的热水萃洗三次(500ml/次),然后减压旋转蒸发掉甲苯(回收有机溶剂甲苯),取丙酮17.6ml加入残留固体中打浆,过滤,滤饼用44.4ml丙酮洗涤一次,滤液收集用于下次套用,转移滤饼至表面皿中烘干,恒重后得到异辛酸铜,绿色固体,约80.9g,收率82.54%,如图1所示,为异辛酸的1hnmr数据谱图,如图2所示,为本发明实施例1制备的异辛酸铜的1hnmr数据谱图。
实施例2
取180.0g异辛酸(1.25mol,2.20equiv)和180.0g水,加入反应器中,搅拌体系。称取45.4gnaoh(1.13mol,2.00equiv)溶于454.0g水中,逐渐滴加至反应器中,然后对体系升温至75℃,并维持此温度2.5h。
取1.62l甲苯加入到反应器中。称取141.8gcuso4·5h2o(0.57mol,1.00equiv)溶于543.5g水中,逐渐滴加至反应器中,滴加完毕后,搅拌反应7.5h。
冷却体系至室温,分液,分出有机相,水相用608ml/次的甲苯萃取两次,合并有机相。有机相用40~50℃的热水萃洗三次(1.0l/次),然后减压蒸馏掉甲苯(回收甲苯),取360ml的丙酮对蒸馏残留物进行打浆,过滤,滤饼用90ml丙酮洗涤一次,滤液收集用于下次套用,转移滤饼至托盘中烘干,恒重后得到异辛酸铜,绿色固体,约170.2g,收率85.34%,核磁数据同实施例1(参见图2)。
实施例3
取500.0g异辛酸(3.47mol,2.20equiv)和180.0g水,加入反应器中,搅拌体系。称取126.1gnaoh(3.15mol,2.00equiv)溶于1.26kg水中,逐渐滴加至反应器中,然后对体系升温至75℃,并维持此温度2.5h。
取4.50l甲苯加入到反应釜中。称取394.0gcuso4·5h2o(1.58mol,1.00equiv)溶于1.51kg水中,逐渐滴加至反应器中,滴加完毕后,搅拌反应7.5h。
冷却体系至室温,分液,分出有机相,水相用1.7l/次的有机溶剂a萃取两次,合并有机相。有机相用40~50℃的热水萃洗三次(2.8l/次),然后减压蒸馏甲苯(回收甲苯),取1.0l丙酮对蒸馏残留物进行打浆,过滤,滤饼用500ml丙酮洗涤一次,滤液收集用于下次套用,转移滤饼至托盘中烘干,恒重后得到异辛酸铜,绿色固体,约458.5g,收率83.14%,核磁数据同实施例1(参见图2)。
实施例4
取18.0g异辛酸(0.125mol,2.20equiv)和18.0g水,加入反应器中,搅拌体系。称取4.5gnaoh(0.113mol,2.00equiv)溶于45.4g水中,逐渐滴加至反应器中,然后对体系升温至50℃,并维持此温度2.5h。
取162.0ml甲苯加入到反应器中。称取14.2gcuso4·5h2o(0.057mol,1.00equiv)溶于54.4g水中,逐渐滴加至反应器中,滴加完毕后,搅拌反应7.5h。
冷却体系至室温,分液,分出有机相,水相用60.8ml/次的甲苯萃取两次,合并有机相。有机相用40~50℃的热水萃洗三次(100.0ml/次),然后减压蒸馏掉甲苯(回收甲苯),取36.0ml的丙酮对蒸馏残留物进行打浆,过滤,滤饼用9.0ml丙酮洗涤一次,滤液收集用于下次套用,转移滤饼至托盘中烘干,恒重后得到异辛酸铜,绿色固体,约13.0g,收率65.21%,核磁数据同实施例1(参见图2)。
实施例5
取18.0g异辛酸(0.125mol,2.20equiv)和18.0g水,加入反应器中,搅拌体系。称取4.5gnaoh(0.113mol,2.00equiv)溶于45.4g水中,逐渐滴加至反应器中,然后对体系升温至100℃,并维持此温度2.5h。
取162.0ml甲苯加入到反应器中。称取14.2gcuso4·5h2o(0.057mol,1.00equiv)溶于54.4g水中,逐渐滴加至反应器中,滴加完毕后,搅拌反应7.5h。
冷却体系至室温,分液,分出有机相,水相用60.8ml/次的甲苯萃取两次,合并有机相。有机相用40~50℃的热水萃洗三次(100.0ml/次),然后减压蒸馏掉甲苯(回收甲苯),取36.0ml的丙酮对蒸馏残留物进行打浆,过滤,滤饼用9.0ml丙酮洗涤一次,滤液收集用于下次套用,转移滤饼至托盘中烘干,恒重后得到异辛酸铜,绿色固体,约15.1g,收率75.81%,核磁数据同实施例1(参见图2)。
实施例6
取18.0g异辛酸(0.125mol,2.20equiv)和18.0g水,加入反应器中,搅拌体系。称取4.5gnaoh(0.113mol,2.00equiv)溶于45.4g水中,逐渐滴加至反应器中,然后对体系升温至75℃,并维持此温度2.5h。
取162.0ml正庚烷加入到反应器中。称取14.2gcuso4·5h2o(0.057mol,1.00equiv)溶于54.4g水中,逐渐滴加至反应器中,滴加完毕后,搅拌反应7.5h。
冷却体系至室温,分液,分出有机相,水相用60.8ml/次的正庚烷萃取两次,合并有机相。有机相用40~50℃的热水萃洗三次(100.0ml/次),然后减压蒸馏掉正庚烷(回收正庚烷),取36.0ml的丙酮对蒸馏残留物进行打浆,过滤,滤饼用9.0ml丙酮洗涤一次,滤液收集用于下次套用,转移滤饼至托盘中烘干,恒重后得到异辛酸铜,绿色固体,约16.9g,收率84.82%,核磁数据同实施例1(参见图2)。
实施例7
取18.0g异辛酸(0.125mol,2.20equiv)和18.0g水,加入反应器中,搅拌体系。称取4.5gnaoh(0.113mol,2.00equiv)溶于45.4g水中,逐渐滴加至反应器中,然后对体系升温至75℃,并维持此温度2.5h。
取162.0ml混合溶剂(正己烷:石油醚:正庚烷=1:1:1)加入到反应器中,温度稳定后回流。称取14.2gcuso4·5h2o(0.057mol,1.00equiv)溶于54.4g水中,逐渐滴加至反应器中,滴加完毕后,搅拌反应7.5h。
冷却体系至室温,分液,分出有机相,水相用60.8ml/次的混合溶剂(正己烷:石油醚:正庚烷=1:1:1)萃取两次,合并有机相。有机相用40~50℃的热水萃洗三次(100.0ml/次),然后减压蒸馏掉溶剂(回收溶剂),取36.0ml的丙酮对蒸馏残留物进行打浆,过滤,滤饼用9.0ml丙酮洗涤一次,滤液收集用于下次套用,转移滤饼至托盘中烘干,恒重后得到异辛酸铜,绿色固体,约16.3g,收率81.77%,核磁数据同实施例1(参见图2)。
实施例8
取10.0g异辛酸(69.34mmol,2.20equiv)和10.0g水,加入反应器中,搅拌体系。称取11.0gna2co3(104.01mmol,1.50equiv)溶于55.0g水中(放出气泡),逐渐滴加至反应器中,然后对体系升温至75℃,并维持此温度2.5h。
取90.0ml甲苯加入到反应器中,温度稳定后回流。称取7.9gcuso4·5h2o(31.56mmol,1.00equiv)溶于30.3g水中,逐渐滴加至反应器中,滴加完毕后,搅拌反应7.5h。
冷却体系至室温,分液,分出有机相,水相用33.8ml/次的甲苯(萃取两次,合并有机相。有机相用40~50℃的热水萃洗三次(55.5ml/次),然后减压蒸馏甲苯(回收甲苯),取20.0ml的丙酮对蒸馏残留物进行打浆,过滤,滤饼用5.0ml丙酮洗涤一次,滤液收集用于下次套用,转移滤饼至托盘中烘干,恒重后得到异辛酸铜,绿色固体,约4.9g,收率44.77%,核磁数据同实施例1(参见图2)。
实施例9
取10.0g异辛酸(69.34mmol,2.20equiv)和50.0g水,加入反应器中,搅拌体系。称取2.5gnah(60%,63.04mmol,2.00equiv)缓慢加至反应器中(反应剧烈),然后对体系升温至75℃,并维持此温度2.5h。
取90.0ml甲苯加入到反应器中,温度稳定后回流。称取7.9gcuso4·5h2o(31.56mmol,1.00equiv)溶于30.3g水中,逐渐滴加至反应器中,滴加完毕后,搅拌反应7.5h。
冷却体系至室温,分液,分出有机相,水相用33.8ml/次的甲苯(萃取两次,合并有机相。有机相用40~50℃的热水萃洗三次(55.5ml/次),然后减压蒸馏甲苯(回收甲苯),取20.0ml的丙酮对蒸馏残留物进行打浆,过滤,滤饼用5.0ml丙酮洗涤一次,滤液收集用于下次套用,转移滤饼至托盘中烘干,恒重后得到异辛酸铜,绿色固体,约8.7g,收率78.79%,核磁数据同实施例1(参见图2)。
实施例10
取10.0g异辛酸(69.34mmol,2.20equiv)和10.0g水,加入反应器中,搅拌体系。称取2.5gnaoh(63.04mmol,2.00equiv)溶于25.2g水中,逐渐滴加至反应器中,然后对体系升温至75℃,并维持此温度2.5h。
取90.0ml甲苯加入到反应器中,温度稳定后回流。称取4.2gcucl2(31.52mol,1.00equiv)溶于30.3g水中,逐渐滴加至反应器中,滴加完毕后,搅拌反应7.5h。
冷却体系至室温,分液,分出有机相,水相用33.8ml/次的甲苯(萃取两次,合并有机相。有机相用40~50℃的热水萃洗三次(55.5ml/次),然后减压蒸馏甲苯(回收甲苯),取20.0ml的丙酮对蒸馏残留物进行打浆,过滤,滤饼用5.0ml丙酮洗涤一次,滤液收集用于下次套用,转移滤饼至托盘中烘干,恒重后得到异辛酸铜,绿色固体,约9.1g,收率82.79%,核磁数据同实施例1(参见图2)。
实施例10
表1反应所需原料和条件及反应产率表
取3.0g的化合物c18h2102p(9.99mmol,1.00equiv)两份,氮气保护下,分别加入两支史莱克管中,氮气保护下,分别加入24.0ml的四氢呋喃(thf),搅拌,然后对两个体系冷却至-60℃以下,编号1和2。
分别取5.5mllda(10.99mmol,1.10equiv)两份,分别加入反应体系1和反应体系2中,过程监控内温-60℃以下。
在-60℃下反应1h,向体系1中加入5.2g本发明制备的异辛酸铜(14.99mmol,1.50equiv),向体系2中加入2.0gcucl2(14.99mmol,1.50equiv)。加完后,约2h后,向两个体系中加水淬灭,分别用二氯甲烷萃取,水相分别用二氯甲烷萃洗至无产物。分别合并两个有机相,蒸馏掉溶剂,柱层析分离:体系1,共分离出类白色固体2.8g,收率93.7%;体系2,共分离出类白色固体2.1g,收率70.8%。
上述对比反应表明,本发明制备的异辛酸铜的催化效率在氧化偶联方面要明显高于氯化铜。
尽管本发明的内容已经通过上述优选实施例作了详细介绍,但应当认识到上述的描述不应被认为是对本发明的限制。在本领域技术人员阅读了上述内容后,对于本发明的多种修改和替代都将是显而易见的。因此,本发明的保护范围应由所附的权利要求来限定。
- 还没有人留言评论。精彩留言会获得点赞!