治疗糖尿病的药物沙格列汀的制备方法与流程
本申请涉及一种治疗糖尿病的药物沙格列汀的制备方法,属于西药合成领域。
背景技术:
近年来糖尿病发病率逐年提高,糖尿病已成为继心血管类疾病恶性肿瘤后又一严重威胁人类健康的非传染性疾病。而糖尿病的ii型发病率占90%,ii型糖尿病的发病机制主要是因为人机体对胰岛素的抗性和胰岛素,细胞的功能缺陷所引发的。临床上用于ii型糖尿病治疗药主要有胰岛素分泌促进剂,磺酰脲类、氯茴苯酸类、胰岛素增敏剂类、胰岛素及胰岛素受体激动剂和二肽基肽酶-4抑制剂。
沙格列汀(saxagliptin),化学名为(1s,3s,5s)-2-[(2s)-2-氨基-2-(3-羟基金刚烷-1-基)乙酰基]-2-氮杂双环[3.1.0]己烷-3-甲腈一水合物,是由百时美施贵宝和阿斯利康制药公司联合开发的一种强效选择性二肽基肽酶-抑制剂,用于ii型糖尿病的治疗,化学结构式如下式所示。
近年来对于沙格列汀用药的需求增加,沙格列汀的制备技术受到广泛关注。目前,沙格列汀的合成方法以化学合成法为主。
沙格列汀的结构是由两个非天然氨基酸的衍生物(s)-(3-羟基金刚烷-1-基)甘氨酸(金刚烷部分)与(1s,3s,5s)-2-氮杂双环[3.1.0]己烷-3-甲腈(环丙基吡咯烷部分)通过酰胺键连接形成的拟肽。沙格列汀的合成采用两片段拼合的方法:分别合成氮杂二环己烷衍生物片段和金刚烷衍生物片段,其中氮杂二环己烷片段为沙格列汀制备的难点。现有的氮杂二环己烷衍生物片段的合成可以以l-焦谷氨酸和5-叔丁基二苯基氧甲基-2-吡咯烷酮为起始原料,而l-焦谷氨酸为原料的方法可行性和实用性高于5-叔丁基二苯基样甲基-2-吡咯烷酮。现有的环丙基吡咯烷部分的合成需要依次经过还原、脱水、水解、酰胺化、环丙烷化以及脱保护成盐等众多步骤,合成路线过长,总体反应收率较低。
另外,对于金刚烷部分的合成,现有技术中报道了以(s)-n-叔丁氧羰基-3-羟基-1-金刚烷甘氨酸为原料,经硫醇活性酯活化,再与(1s,3s,5s)-2-氮杂双环[3.1.0]己烷-3-甲腈对甲苯磺酸盐发生缩合反应,经盐酸脱氨基保护基等步骤得到沙格列汀。该过程工艺简单,收率高,适合工业化生产。但硫醇活性酯价格较贵,导致整体成本高。
为解决上述技术问题,本申请提供一种新的制备沙格列汀的方法。基于有机合成领域中金属化合物与有机膦配体在小分子化合物合成方面的应用,通过不断筛选反应条件,发现在特定有机膦配体和钯催化剂存在下,以丁炔二酸二乙酯和氨基保护的(s)-3-氨基-3-氰基-丙酸乙酯为起始原料,经环合、脱羧、环丙烷化,能够高产率、高选择性获得中间产物(1s,3s,5s)-2-氮杂双环[3.1.0]己烷-3-甲腈对甲苯磺酸盐,再与n-羟基琥珀酰亚胺活化的(s)-n-叔丁氧羰基-(3-羟基金刚烷-1-基)甘氨酸反应,提高反应速率,得到高产率高纯度的沙格列汀产物,反应路线大大缩短,各步骤产率高,反应时间短,降低了生产成本,有利于工业化生产。
技术实现要素:
本发明要解决的技术问题是针对现有技术中,沙格列汀反应路线较为繁琐,部分反应条件较为苛刻,反应路线长,原料价格相对昂贵,且产率低,产物纯度不高等问题。
为了解决上述技术问题,本发明提供的技术方案为:
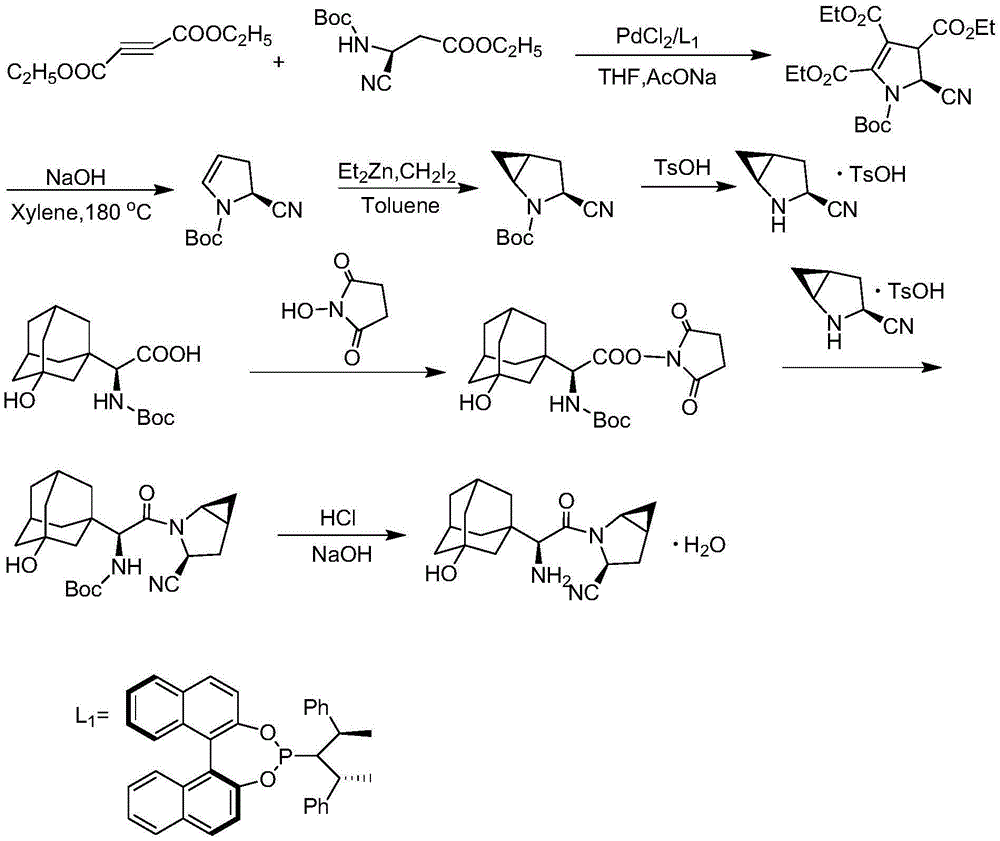
一种沙格列汀的合成方法,其合成路线如下:
具体反应过程包括:
在四氢呋喃中,惰性气体保护下,加入丁炔二酸二乙酯和(s)-n-boc-3-氨基-3-氰基-丙酸乙酯,在催化剂pdcl2和手性配体l1及弱碱醋酸钠存在下,ag2o作为氧化剂,30-50℃下搅拌3-5小时,tlc监测反应进程,待反应完毕,加水分层,有机相无水硫酸镁干燥过夜,减压浓缩,经柱层析,获得环化化合物;
惰性气体选自氮气或氩气;催化剂pdcl2和手性配体l1的摩尔用量是丁炔二酸二乙酯的0.5-5%;醋酸钠的摩尔用量是丁炔二酸二乙酯的2-5倍;ag2o的摩尔用量是丁炔二酸二乙酯的1-3倍;丁炔二酸二乙酯和(s)-n-boc-3-氨基-3-氰基-丙酸乙酯的摩尔用量比为1-2:1。
环化化合物在二甲苯中,加入固体氢氧化钠,氮气保护下封管加热至180℃,反应10-12小时,冷至室温,过滤,减压除去溶剂,经柱层析,获得化合物(s)-n-boc-2-氰基-2,3-二氢-1h-吡咯;
氢氧化钠的用量是环化化合物质量的1-5倍。
在甲苯中,氮气保护下,加入(s)-n-boc-2-氰基-2,3-二氢-1h-吡咯,冷却至-40℃--20℃,滴加新制备的二乙基锌溶液,保温搅拌0.5-1小时,滴加二碘甲烷的甲苯溶液,滴加完毕,自然升温至室温,继续反应2-5小时,加入冰水淬灭反应,分液,有机相浓缩,经柱层析获得环丙烷化产物;
二乙基锌的摩尔用量是(s)-n-boc-2-氰基-2,3-二氢-1h-吡咯的2-3倍,二碘甲烷的摩尔用量是(s)-n-boc-2-氰基-2,3-二氢-1h-吡咯的1-2倍。
在二氯甲烷中,加入环丙烷化产物,加入1-2摩尔量的对甲苯磺酸,回流反应2-3小时,冷至室温,过滤,得(1s,3s,5s)-2-氮杂双环[3.1.0]己烷-3-甲腈对甲苯磺酸盐。
在二氯甲烷中,加入(s)-n-boc-(3-羟基金刚烷-1-基)甘氨酸和n-羟基琥珀酰亚胺、三乙胺,20℃-40℃下搅拌反应0.5-1小时,加水分层,有机相减压浓缩,经柱层析,获得(s)-n-boc-(3-羟基金刚烷-1-基)甘氨酸琥珀酰亚胺酯;
(s)-n-boc-(3-羟基金刚烷-1-基)甘氨酸和n-羟基琥珀酰亚胺、三乙胺的摩尔用量比为1:1-2:1-3。
在四氢呋喃中,加入(s)-n-boc-(3-羟基金刚烷-1-基)甘氨酸琥珀酰亚胺酯和(1s,3s,5s)-2-氮杂双环[3.1.0]己烷-3-甲腈对甲苯磺酸盐、三乙胺,室温反应1-2小时,盐酸调节ph至中性,分液,有机相减压浓缩,经柱层析,获得产物(1s)-2-[(1s,3s,5s)-3-氰基-2-氮杂双环[3.1.0]己烷-2-基]-1-(3-羟基金刚烷-1-基)-2-氧代乙基氨基甲酸叔丁酯;
(s)-n-boc-(3-羟基金刚烷-1-基)甘氨酸琥珀酰亚胺酯和(1s,3s,5s)-2-氮杂双环[3.1.0]己烷-3-甲腈对甲苯磺酸盐、三乙胺的摩尔用量比为1:1-2:2-4。
在异丙醇中,加入(1s)-2-[(1s,3s,5s)-3-氰基-2-氮杂双环[3.1.0]己烷-2-基]-1-(3-羟基金刚烷-1-基)-2-氧代乙基氨基甲酸叔丁酯,加热至60-80℃,滴加盐酸,继续反应2-3小时,冷至室温,滴加氢氧化钠水溶液至ph到9-10,加入二氯甲烷,继续搅拌20-30分钟,分液,有机相减压浓缩,经柱层析,获得沙格列汀一水合物。
本发明的有益效果为:
本发明提供了一种全新的制备沙格列汀的合成路线,反应路线大大缩短,采用的原料更加廉价易得,反应过程容易操作,各步骤产率高,成环反应具有大于99.9%e.e.值的手性选择性,通过对羧基的活化,反应选择性更强,副产物减少,所获得产物纯度高,降低了生产成本,有利于工业化生产。
附图说明
图1为沙格列汀的合成路线。
具体实施方式
本发明公开了一种沙格列汀的制备方法,本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。需要特别指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明,并且相关人员明显能在不脱离本发明内容、精神和范围的基础上对本文所述内容进行改动或适当变更与组合,来实现和应用本发明技术。
在本发明中,除非另有说明,否则本文中使用的科学和技术名词具有本领域技术人员所通常理解的含义。
为了使本领域的技术人员更好地理解本发明的技术方案,下面结合具体实施例对本发明作进一步的详细说明。
实施例1:环化化合物的制备
在120ml四氢呋喃中,氮气保护下,加入7.65g(45mmol)丁炔二酸二乙酯和10.91g(45mmol)(s)-n-boc-3-氨基-3-氰基-丙酸乙酯,0.22g(1.2mmol)pdcl2和0.65g(1.2mmol)手性配体l1及8.21g(100mmol)醋酸钠,18.53g(80mmol)ag2o作为氧化剂,40℃下搅拌3小时,tlc监测反应进程,待反应完毕,加水分层,有机相无水硫酸镁干燥过夜,减压浓缩,经柱层析,获得环化化合物17.93g(43.7mmol),产率为97%,e.e.>99.9%。
1hnmr(chcl3-d,400m)δ:5.25(d,1h),4.26(m,2h),4.15(m,2h),4.05(m,2h),3.67(d,1h),1.55(s,9h),1.24(m,3h),1.19(m,3h),1.11(m,3h);13cnmr(chcl3-d,400m)δ:171.1,167.8,165.1,148.2,135.5,122.1,118.9,87.6,67.1,61.6,43.8,38.5,26.9,14.1;ms-esi(m/z):411.12[m+h]+。
实施例2:(s)-n-boc-2-氰基-2,3-二氢-1h-吡咯的制备
在150mlschlenk反应管中,加入4.10g(10mmol)环化化合物和100ml二甲苯,固体氢氧化钠6.33g,氮气保护,封管加热至180℃,反应10小时,冷至室温,过滤,水洗,减压除去溶剂,经柱层析,获得化合物(s)-n-boc-2-氰基-2,3-二氢-1h-吡咯1.57g(8.1mmol),产率为81%。
实施例3:(1s,3s,5s)-2-氮杂双环[3.1.0]己烷-3-甲腈的制备
在120ml甲苯中,氮气保护下,加入4.86g(25mmol)(s)-n-boc-2-氰基-2,3-二氢-1h-吡咯,冷却至-30℃,滴加新制备的二乙基锌溶液22ml(约含55mmol二乙基锌),保温搅拌0.5小时,滴加二碘甲烷的甲苯溶液30ml(约含30mmol二碘甲烷),滴加完毕,自然升温至室温,继续反应3小时,加入冰水淬灭反应,分液,有机相浓缩,经柱层析,获得环丙烷化产物3.96g(19mmol),产率为76%。
实施例4:(1s,3s,5s)-2-氮杂双环[3.1.0]己烷-3-甲腈对甲苯磺酸盐的制备
在100ml二氯甲烷中,加入10.42g(50mmol)环丙烷化产物,9.47g(55mmol)对甲苯磺酸,回流反应2小时,冷至室温,浓缩,滴加乙醚,固体析出,过滤,得(1s,3s,5s)-2-氮杂双环[3.1.0]己烷-3-甲腈对甲苯磺酸盐13.62g(48.6mmol),产率为97%。
实施例5:(s)-n-boc-(3-羟基金刚烷-1-基)甘氨酸琥珀酰亚胺酯的制备
在150ml二氯甲烷中,加入8.14g(25mmol)(s)-n-boc-(3-羟基金刚烷-1-基)甘氨酸和3.45g(30mmol)n-羟基琥珀酰亚胺、5.04g(50mmol)三乙胺,25℃下搅拌反应1小时,盐酸调节ph至7.5,加水分层,有机相减压浓缩,经柱层析,获得(s)-n-boc-(3-羟基金刚烷-1-基)甘氨酸琥珀酰亚胺酯9.55g(22.6mmol),产率为90%。
1hnmr(chcl3-d,400m)δ:4.15(s,1h),2.69(m,4h),1.13-1.42(m,23h);13cnmr(chcl3-d,400m)δ:176.1,169.8,157.1,79.2,69.9,55.1,45.7,38.6,37.1,29.6,28.4,25.5,19.1;ms-esi(m/z):423.22[m+h]+。
实施例6:(1s)-2-[(1s,3s,5s)-3-氰基-2-氮杂双环[3.1.0]己烷-2-基]-1-(3-羟基金刚烷-1-基)-2-氧代乙基氨基甲酸叔丁酯的制备
在150ml四氢呋喃中,加入6.35g(15mmol)(s)-n-boc-(3-羟基金刚烷-1-基)甘氨酸琥珀酰亚胺酯和4.21g(15mmol)(1s,3s,5s)-2-氮杂双环[3.1.0]己烷-3-甲腈对甲苯磺酸盐、3.34g(33mmol)三乙胺,室温反应2小时,盐酸调节ph至中性,分液,有机相减压浓缩,经柱层析,获得产物(1s)-2-[(1s,3s,5s)-3-氰基-2-氮杂双环[3.1.0]己烷-2-基]-1-(3-羟基金刚烷-1-基)-2-氧代乙基氨基甲酸叔丁酯4.82g(11.6mmol),产率为77%。
实施例7:沙格列汀的制备
在100ml异丙醇中,加入8.32g(20mmol)(1s)-2-[(1s,3s,5s)-3-氰基-2-氮杂双环[3.1.0]己烷-2-基]-1-(3-羟基金刚烷-1-基)-2-氧代乙基氨基甲酸叔丁酯,加热至60℃,滴加盐酸,至ph为2.5,继续反应2小时,冷至室温,滴加氢氧化钠水溶液至ph为10,加入100ml二氯甲烷,继续搅拌30分钟,分液,水相以二氯甲烷萃取一次,合并有机相,有机相减压浓缩,经柱层析,获得沙格列汀一水合物5.21g(15.6mmol),产率为78%,hplcpurity=99.78%。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!