一种制备双丙酸阿氯米松用的还原后中间体产品的方法与流程
本发明属于甾体激素药物的制备工艺技术,具体涉及到一种制备双丙酸阿氯米松用的还原后中间体产品的方法。
背景技术:
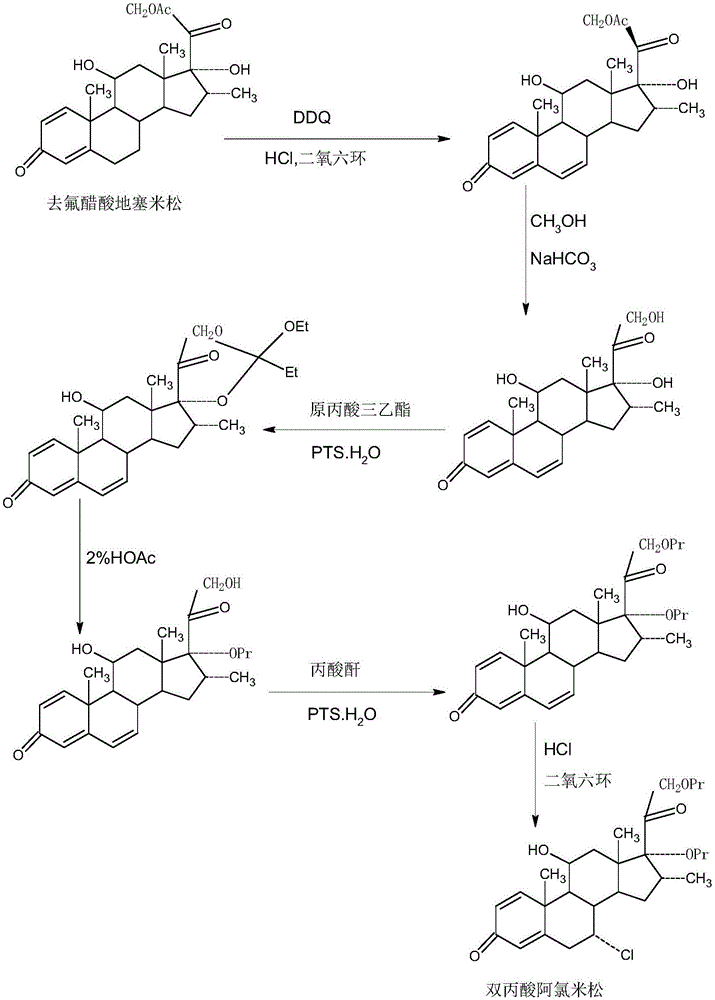
双丙酸阿氯米松(分子式c28h37clo7),化学名为7a-氯-16a-甲基-11-羟基-孕甾-1,4-二烯-3,20-二酮-17,21-双丙酸酯,是一种强效甾体皮质激素药物,有强效的局部抗炎、止痒及收缩血管的作用,在临床上主要用于湿疹、特应性皮炎、牛皮癣、及其它皮肤病的治疗,副作用低,效果好,市场前景广阔。双丙酸阿氯米松的传统生产方法,是以去氟醋酸地塞米松为原料,经ddq6位脱氢,21位碱催化水解,17,21位原丙酸三乙酯环酯化,酸催化环酯水解,21位丙酯化,6,7位hcl气体加成上7a-cl等六步化学反应,制得双丙酸阿氯米松,其工艺路线见附图1。该合成方法,合成路线长,合成总收率只有2.678%,尤其是最后一步6,7位hcl气体加成反应,合成收率不到20%,由于合成总收率低,多步合成化学反应副反应多,产生的杂质量大,精制提纯难,同时产生三废也较多,且不易处理,易污染环境,使得双丙酸阿氯米松生产成本与市场价格均很高,加重了该类病人的用药负担。
因此,本领域需要一种新的双丙酸阿氯米松的制备方法。
技术实现要素:
本发明提供一种制备双丙酸阿氯米松用的还原后中间体产品的方法。
本发明的技术方案是,一种制备双丙酸阿氯米松用的还原后中间体产品的方法,所述方法包括以16a-甲基表氢化可的松为原料,经21位丙酸酯化,7,11位双氧化成双酮,17位丙酸酯化,3位烯醇化醚化保护,7,11位双酮还原并酸水解脱保护共五步反应合成双丙酸阿氯米松用的还原后中间体粗品,具体步骤包括:
a、合成酯化物,是以16a-甲基表氢化可的松为原料,将其在第一有机溶剂中与丙酸酐在酸催化剂催化下,21位的羟基发生酯化反应,得酯化物:16a-甲基表氢化可的松-21丙酸酯;
b、合成氧化物,是将酯化物在含酸的第二有机溶剂中与氧化剂跟分子中11a羟基与7位活泼氢发生7,11位氧化反应,得氧化物:16a-甲基-17a-羟基-21-丙酰氧基-孕甾-4烯-
c、合成双酯物,是将氧化物在第三有机溶剂中与丙酸酐在酸催化剂催化下,17a-羟基发生酯化反应生成双酯物:16a-甲基-17a,21-双丙酰氧基-孕甾-4-烯-3,7,11,20-四酮;
d、合成醚化物,是将双酯物在第四有机溶剂中与原甲酸三乙酯在酸催化剂催化下跟分子中的3-酮-4-烯结构中的酮基发生烯醇式醚化保护反应,得醚化物:3-乙烯醇醚基-16a-甲基-17a,21-双丙酰氧基-孕甾-3,5-二烯-7,11,20-三酮;
e、合成还原物,是将醚化物在第五有机溶剂中与还原剂跟分子中7,11位的酮基发生还原反应,并在酸催化下脱保护,得还原物即双丙酸阿氯米松用的还原后中间体粗品;
再对该双丙酸阿氯米松用的还原后中间体粗品进行重结晶精制得到双丙酸阿氯米松用的还原后中间体产品。
进一步地,所述方法的具体操作步骤如下:
a、合成酯化物:是将16a-甲基表氢化可的松溶解于第一有机溶剂中,于10~50℃下加入丙酸酐,酸催化剂催化下反应6~8小时,tlc确认反应终点,反应完后,加碱中和,减压回收有机溶剂,然后以乙醇水溶液结晶,干燥得酯化物:16a-甲基表氢化可的松-21丙酸酯,hplc含量97.0~98.5%,重量收率110~115%;
b、合成氧化物:是将上述酯化物溶入第二有机溶剂中,在10~50℃下,2~3小时内慢慢滴加氧化剂,滴完后再保温于10~50℃继续反应1~2小时,tlc确认反应终点,反应完后,加入适量的保险粉以除去过量的氧化剂,减压浓缩回收90~95%有机溶剂,然后降温水析,离心,滤液排入废水处理池,滤饼干燥,得氧化物,hplc含量95.0~98.5%,重量收率95~100%;
c、合成双酯物:是将上述氧化物溶入第三有机溶剂中,保温于10~80℃下加入丙酸酐,酸催化剂催化下反应6~8小时,tlc确认反应终点,反应完后,降温至25~30℃,加入适量的碱中和至溶液的ph6.5~7.5,减压浓缩回收90~95%的有机溶剂后,冷却降温至10~25℃,加入纯水,搅拌析晶1.5~2.5小时,离心,滤液排入废水处理池,滤饼洗涤干燥,得双酯物:16a-甲基-17a,21-双丙酰氧基-孕甾-4-烯-3,7,11,20-四酮,hplc含量98.0~99.0%,重量收率110~115%;
d、合成醚化物:是将上述双酯物溶入第四有机溶剂中,加入原甲酸三乙酯和酸催化剂,保温于10~80℃反应3~4小时,tlc确认反应终点,反应完后,加入适量的碱中和至溶液的ph6.5~7.5,减压浓缩回收90~95%的有机溶剂后,冷却降温至10~25℃,加入纯水,搅拌析晶1.5~2.5小时,离心,滤液排入废水处理池,滤饼洗涤干燥,得醚化物:3-乙烯醇醚基-16a-甲基-17a,21-双丙酰氧基-孕甾-3,5-二烯-7,11,20-三酮,hplc含量97.0~99.0%,重量收率100~105%;
e、合成还原物:是将上述醚化物溶解在第五有机溶剂中,保温于10~80℃下,慢慢加入还原剂,1.5~2.5小时加完,然后再于10~80℃继续保温反应1~2小时,tlc确认反应终点,反应完后,慢慢滴加适量的酸催化剂水溶液,使体系ph至2~3,再于10~80℃继续水解反应2~3小时,tlc确认反应终点,反应完后,慢慢加适量碱中和至体系ph至6~7,然后减压浓缩回收90~95%有机溶剂,再降温,慢慢加入纯水水析,离心,滤液排入废水处理池,滤饼干燥,得双丙酸阿氯米松用的还原后中间体粗品,将该粗品用酒精水溶液重结晶,得双丙酸阿氯米松用的还原后中间体产品:16a-甲基-7,11-二羟基-17a,21-双丙酰氧基-孕甾-4-烯-3,20-二酮,hplc含量98.0~99.0%,重量收率70~75%。
进一步地,所述具体步骤中:
a、合成酯化物中所述第一有机溶剂是丙酸酐、甲苯、丙酸、氯仿、dme、二氧六环中的一种;反应的酸催化剂是无机强酸硫酸、盐酸或有机强酸对甲苯磺酸中的一种;反应物间的重量配比是,16a-甲基表氢化可的松:丙酸酐:酸催化剂=1g:0.3~0.5g:0.01~0.15g;反应物与溶剂间的配比是,16a-甲基表氢化可的松:第一有机溶剂=1g:1~5ml;
b、合成氧化物中所述第二有机溶剂是甲苯、丙酮、氯仿、dme、四氢呋喃、dmso、二氧六环、冰醋酸中的一种;所述氧化剂是氧化铬、ppc、次卤酸钠中的一种;反应物间的重量配比是,酯化物:氧化剂=1g:0.2~0.8g;反应物与溶剂间的配比是,酯化物:第二有机溶剂=1g:3~10ml;
c、合成双酯物中所述第三有机溶剂是丙酸酐、甲苯、丙酸、氯仿、dme、二氧六环中的一种;反应的酸催化剂是无机强酸硫酸、盐酸或有机强酸对甲苯磺酸中的一种;反应物间的重量配比是,氧化物:丙酸酐:酸催化剂=1g:0.3~0.5g:0.05~0.25g;反应物与溶剂间的配比是,氧化物:第三有机溶剂=1g:1~8ml;
d、合成醚化物中所述第四有机溶剂是甲苯、二氯甲烷、氯仿、四氢呋喃、二氧六环、dme、c4以下低级醇中的一种;反应的酸催化剂是无机强酸硫酸、盐酸或有机强酸对甲苯磺酸、三氟乙酸中的一种;反应物间的重量配比是,双酯物:原甲酸三乙酯:酸催化剂=1g:0.6~1.2ml:0.05~0.25g;反应物与溶剂间的配比是,双酯物:第四有机溶剂=1g:2~8ml。
进一步地,所述具体步骤中:
a、合成酯化物中所述第一有机溶剂是丙酸;反应温度为25~30℃;反应的酸催化剂是对甲苯磺酸;反应物间的重量配比是,16a-甲基表氢化可的松:丙酸酐:酸催化剂=1g:0.4g:0.05g;反应物与溶剂间的配比是,16a-甲基表氢化可的松:第一有机溶剂=1g:2ml;
b、合成氧化物中所述第二有机溶剂是冰醋酸;反应温度为20~25℃;所述氧化剂是氧化铬;反应物间的重量配比是,酯化物:氧化剂=1g:0.4g;反应物与溶剂间的配比是,酯化物:第二有机溶剂=1g:5ml;
c、合成双酯物中所述第三有机溶剂是丙酸;反应温度为30~35℃;反应的酸催化剂是对甲苯磺酸;反应物间的重量配比是,氧化物:丙酸酐:酸催化剂=1g:0.4g:0.1g;反应物与溶剂间的配比是,氧化物:第三有机溶剂=1g:3ml;
d、合成醚化物中所述第四有机溶剂是二氯甲烷;反应的酸催化剂是对甲苯磺酸;反应温度为20~25℃;反应物间的重量配比是,双酯物:原甲酸三乙酯:酸催化剂=1g:0.9ml:0.10g;反应物与溶剂间的配比是,双酯物:第四有机溶剂=1g:6ml。
进一步地,所述具体步骤中:
d、合成醚化物中中和酸的碱是无机碱氢氧化钠、碳酸钠或有机碱吡啶、三乙胺中的一种;
e、合成还原物中水解反应的酸催化剂是无机强酸硫酸、盐酸或有机强酸对甲苯磺酸中的一种。
进一步地,所述具体步骤中:
d、合成醚化物中中和酸的碱是三乙胺;
e、合成还原物中水解反应的酸催化剂是盐酸。
进一步地,所述具体步骤中:
e、合成还原物中所述第五有机溶剂是甲苯、二氯甲烷、氯仿、四氢呋喃、c4以下低碳醇中的一种;所述还原剂是硼氢化钠、硼氢化钾、硼氢化钙中的一种;还原反应物间的重量配比是,醚化物:还原剂=1g:0.12~0.24g;水解反应物间的重量配比是,醚化物:酸催化剂=1g:0.2~0.5g;反应物与溶剂间的配比是,醚化物:第五有机溶剂=1g:8~16ml。
进一步地,所述具体步骤中:
e、合成还原物中所述第五有机溶剂是甲醇;所述还原剂是硼氢化钠;还原反应和水解反应的温度均为25~30℃;还原反应物间的重量配比是,醚化物:还原剂=1g:0.16g;水解反应物间的重量配比是,醚化物:酸催化剂=1g:0.3g;反应物与溶剂间的配比是,醚化物:第五有机溶剂=1g:12m。
本发明的有益效果包括:
本发明以16a-甲基表氢化可的松为原料,经酯化、氧化、双酯化、醚化、还原等五步反应合成双丙酸阿氯米松用的还原后中间体,本发明工艺具有合成路线短,工艺经济环保,生产操作简便,产品收率高等诸多优点;生产中使用的溶剂,可回收循环套用,易实施工业化生产。
附图说明
图1为传统双丙酸阿氯米松合成路线图。
图2为本发明双丙酸阿氯米松合成路线图。
具体实施方式
为了更详细地说明本发明的要点与精神,下面举几个实施例予以说明。
实施例1
a、酯化物的制备:在一个1000ml三口瓶中,加入100g16a-甲基表氢化可的松,200ml丙酸,40g丙酸酐,5g对甲苯磺酸,于20~25℃反应6~8小时,tlc确认反应终点,反应完后,将反应液慢慢滴加至装有2000ml摩尔浓度为5%氢氧化钠溶液的5000ml三口瓶中,于0~5℃搅拌析晶4~5小时,过滤,滤液排入废水处理池,滤饼加入至600ml摩尔浓度为20%的乙醇水溶液中,于30~35℃下搅拌5~6小时,再降温至0~5℃搅拌析晶5~6小时,过滤,滤液套用于下批精制工艺,滤饼洗涤,干燥,得酯化物:16a-甲基表氢化可的松-21丙酸酯112g,hplc含量97.4%,重量收率112%;
b、氧化物的制备:在一个1000ml三口瓶中,加入100g上述a所制备的酯化物,500ml冰醋酸,搅拌使之全溶,然后控温20~25℃下,慢慢滴加40g氧化铬与80g水配成的氧化剂溶液,约2.5~3小时内滴完,滴完后再保温于20~25℃继续反应1~2小时,tlc确定反应终点,反应完后,加入10g的保险粉以除去过量的氧化剂,然后减压浓缩回收约90%的冰醋酸,再降温至20~30℃,加入500ml纯水,于0~5℃搅拌析晶3~4小时,过滤,滤液排入废水处理池,滤饼洗涤干燥,得氧化物98g,hplc含量97.5%,重量收率98%;
c、双酯物的制备:在一个1000ml三口瓶中,加入100g上述b所制备的氧化物,300ml丙酸,40g丙酸酐,10g对甲苯磺酸,保温于30~35℃下反应6~8小时,tlc确认反应终点,反应完后,降温至25~30℃,将反应液慢慢滴加至装有2000ml摩尔浓度为5%氢氧化钠溶液的5000ml三口瓶中,于0~5℃搅拌析晶4~5小时,过滤,滤液排入废水处理池,滤饼水洗,干燥,得双酯物:16a-甲基-17a,21-双丙酰氧基-孕甾-4烯-3,7,11,20-四酮113.6g,hplc含量98.5%,重量收率113.6%;
d、醚化物的制备:在一个1000ml三口瓶中,加入100g上述c所制备的双酯物,600ml二氯甲烷后,90ml原甲酸三乙酯和10g对甲苯磺酸,保温于20~25℃反应3~4小时,tlc确认反应终点,反应完后,加入适量的三乙胺中和至溶液的ph为6.5~7.5,减压浓缩回收90~95%的二氯甲烷后,冷却降温至20~25℃,加入500ml纯水,于0~5℃搅拌析晶1.5~2.5小时,过滤,滤液排入废水处理池,滤饼洗涤干燥,得醚化物:3-乙烯醇醚基-16a-甲基-17a,21-双丙酰氧基-孕甾-3,5-二烯-7,11,20-三酮104g,hplc含量97.6%,重量收率104%;
e、还原物的制备:在一个2000ml三口瓶中,加入100g上述d所制备的醚化物,1200ml甲醇,保温于25~30℃下,搅拌使固体全部溶解,再慢慢加入16g硼氢化钠还原剂,约1.5~2.5小时加完,然后再于25~30℃继续保温反应1~2小时,tlc确认反应终点,反应完后,慢慢滴加100g摩尔浓度为30%的盐酸水溶液,使体系ph至2~3,再于25~30℃继续水解反应2~3小时,tlc确认反应终点,反应完后,慢慢加适量碱中和至体系ph至6~7,然后减压浓缩回收90~95%有机溶剂甲醇,再降温,慢慢加入600ml纯水水析,过滤,滤液排入废水处理池,滤饼洗涤干燥,得还原物粗品,将该粗品用500ml摩尔浓度为50%的酒精水溶液重结晶,得还原物:16a-甲基-7,11-二羟基-17a,21-双丙酰氧基-孕甾-4-烯-3,20-二酮74.2g,hplc含量98.8%,重量收率74.2%;
f、1-脱氢物的制备:在一个2000ml三口瓶中,加入100g上述e所制备的还原物,1200ml二氧六环,80gddq脱氢剂,搅拌使其完全溶解,升温至75~80℃保温反应6~10小时,tlc确认反应终点,反应完后,滤除反应生成的氢醌,再加入适量的亚硫酸氢钠溶液破坏过量的ddq,然后减压浓缩回收90~95%有机溶剂二氧六环,再降温,慢慢加入600ml纯水水析,离心,滤液排入废水处理池,滤饼洗涤干燥,得1-脱氢物粗品,将该粗品用500ml含20%酒精10%烧碱的水溶液重结晶,得1-脱氢物:16a-甲基-7,11-二羟基-17a,21-双丙酰氧基-孕甾-1,4-二烯-3,20-二酮63.8g,hplc含量98.2%,重量收率63.8%;
g、双丙酸阿氯米松的制备:在一个2000ml三口瓶中,加入100g上述f所制备的1-脱氢物,800ml甲苯,200g吡啶,升温搅拌使固体全部溶解后,再加入80g三氯化磷氯化剂,保温于40~45℃搅拌反应4~6小时,tlc确认反应终点,反应完后,加入500g摩尔浓度为30%的烧碱溶液分2次中和洗涤至水溶液层ph为6~7,减压浓缩回收90~95%的有机溶剂甲苯后,冷却降温至20~25℃,加入600ml纯水,于0~5℃搅拌析晶1.5~2.5小时,过滤,滤液排入废水处理池,滤饼洗涤干燥,得双丙酸阿氯米松粗品,将该粗品用600ml酒精与5g活性炭回流脱色1小时,过滤,滤饼洗涤,滤液和洗液合并浓缩80%的酒精,于0~5℃搅拌结晶3~4小时,处理所得固体再用甲醇-丙酮-异丙醚=2:1:3的混合溶剂重结晶,得双丙酸阿氯米松:7a-氯-16a-甲基-11-羟基-孕甾-1,4-二烯-3,20-二酮-17,21-双丙酸酯64.2g,熔点:214~216℃,hplc含量99.4%,重量收率64.2%。
实施例2
a、酯化物的制备:在一个1000ml三口瓶中,加入100g16a-甲基表氢化可的松,500ml氯仿,40g丙酸酐,8g对甲苯磺酸,于20~25℃反应6~8小时,tlc确认反应终点,反应完后,慢慢滴加20ml摩尔浓度为50%氢氧化钠溶液,分水后减压浓缩回收90~95%的氯仿,然后加入500ml纯水,于0~5℃搅拌析晶4~5小时,过滤,滤液排入废水处理池,滤饼加入至600ml摩尔浓度为20%的乙醇水溶液中,于30~35℃下搅拌5~6小时,再降温至0~5℃搅拌析晶5~6小时,过滤,滤液套用于下批精制工艺,滤饼洗涤,干燥,得酯化物:16a-甲基-表氢化可的松-21丙酸酯109.5g,hplc含量97.8%,重量收率109.5%;
b、氧化物的制备:在一个1000ml三口瓶中,加入100g上述a所制备的酯化物,600ml丙酮,搅拌使之全溶,然后控温20~25℃下,慢慢滴加40g氧化铬与80g纯水配成的氧化剂溶液,约2.5~3小时内滴完,滴完后再保温于20~25℃继续反应1~2小时,tlc确认反应终点,反应完后,加入10g的保险粉以除去过量的氧化剂,然后减压浓缩回收约90%的丙酮,再降温至20~30℃,加入500ml纯水,于0~5℃搅拌析晶3~4小时,过滤,滤液排入废水处理池,滤饼洗涤干燥,得氧化物97.4g,hplc含量96.6%,重量收率97.4%;
c、双酯物的制备:在一个1000ml三口瓶中,加入100g上述b所制备的氧化物,300ml氯仿,40g丙酸酐,10g对甲苯磺酸,保温于50~55℃下反应6~8小时,tlc确认反应终点,反应完后,降温至25~30℃,向反应液中慢慢滴加至50ml摩尔浓度为30%氢氧化钠溶液,分去水层,再用50ml纯水洗涤一次,分尽水,然后减压浓缩回收90~95%的溶剂氯仿,冷却后加入500ml纯水,于0~5℃搅拌析晶4~5小时,过滤,滤液排入废水处理池,滤饼水洗,干燥,得双酯物:16a-甲基-17a,21-双丙酰氧基-孕甾-4-烯-3,7,11,20-四酮112.2g,hplc含量97.8%,重量收率112.2%;
d、醚化物的制备:在一个1000ml三口瓶中,加入100g上述c所制备的双酯物,600mlthf,90ml原甲酸三乙酯和10g浓硫酸,保温于20~25℃反应3~4小时,tlc确认反应终点,反应完后,加入适量的碳酸钠固体中和至溶液的ph6.5~7.5,减压浓缩回收90~95%的thf后,冷却降温至20~25℃,加入500ml纯水,于0~5℃搅拌析晶1.5~2.5小时,过滤,滤液排入废水处理池,滤饼洗涤干燥,得醚化物:3-乙烯醇醚基-16a-甲基-17a,21-双丙酰氧基-孕甾-3,5-二烯-7,11,20-三酮101.8g,hplc含量96.4%,重量收率101.8%;
e、还原物的制备:在一个2000ml三口瓶中,加入100g上述d所制备的醚化物,1200mldme,保温于25~30℃下,搅拌使固体全部溶解,再慢慢加入18g硼氢化钠还原剂,约1.5~2.5小时加完,然后再于25~30℃继续保温反应1~2小时,tlc确认反应终点,反应完后,慢慢滴加120g摩尔浓度为30%的盐酸水溶液,使体系ph至2~3,再于25~30℃继续水解反应2~3小时,tlc确认反应终点,反应完后,慢慢加适量碱中和至体系ph至6~7,然后减压浓缩回收90~95%有机溶剂dme,再降温,慢慢加入600ml纯水水析,过滤,滤液排入废水处理池,滤饼洗涤干燥,得还原物粗品,将该粗品用500ml摩尔浓度为50%的酒精水溶液重结晶,得还原物:16a-甲基-7,11-二羟基-17a,21-双丙酰氧基-孕甾-4-烯-3,20-二酮72.6g,hplc含量97.2%,重量收率72.6%;
f、1-脱氢物的制备:在一个2000ml三口瓶中,加入100g上述e所制备的还原物,1200ml甲苯,80gddq脱氢剂,搅拌使其完全溶解,升温至75~80℃保温反应6~10小时,tlc确认反应终点,反应完后,滤除反应生成的氢醌,再加入适量的亚硫酸氢钠溶液破坏过量的ddq,然后减压浓缩回收90~95%有机溶剂甲苯,再降温,慢慢加入600ml纯水水析,离心,滤液排入废水处理池,滤饼洗涤干燥,得1-脱氢物粗品,将该粗品用500ml含20%酒精10%烧碱的水溶液重结晶,得1-脱氢物:16a-甲基-7,11-二羟基-17a,21-双丙酰氧基-孕甾-1,4-二烯-3,20-二酮61.5g,hplc含量97.8%,重量收率61.5%;
g、双丙酸阿氯米松的制备:在一个2000ml三口瓶中,加入100g上述f所制备的1-脱氢物,800ml氯仿,200g三乙胺,升温搅拌使固体全部溶解后,再加入80g三氯化磷氯化剂,保温于40~45℃搅拌反应4~6小时,tlc确认反应终点,反应完后,加入500g摩尔浓度为30%的烧碱溶液分2次中和洗涤至水溶液层ph6~7,减压浓缩回收90~95%的有机溶剂氯仿后,冷却降温至20~25℃,加入600ml纯水,于0~5℃搅拌析晶1.5~2.5小时,过滤,滤液排入废水处理池,滤饼洗涤干燥,得双丙酸阿氯米松粗品,将该粗品按实施例1中g所述方法重结晶,得双丙酸阿氯米松:7a-氯-16a-甲基-11-羟基-孕甾-1,4-二烯-3,20-二酮-17,21-双丙酸酯61.6g,熔点:214~215.5℃,hplc含量99.3%,重量收率61.6%。
实施例3
a、酯化物的制备:在一个1000ml三口瓶中,加入100g16a-甲基表氢化可的松,500ml甲苯,40g丙酸酐,6g三氟乙酸,于20~25℃反应6~8小时,tlc确认反应终点,反应完后,慢慢滴加20ml摩尔浓度为50%氢氧化钠溶液,分水后减压浓缩回收90~95%的甲苯,然后加入500ml纯水,于0~5℃搅拌析晶4~5小时,过滤,滤液排入废水处理池,滤饼加入至600ml摩尔浓度为20%的乙醇水溶液中,于30~35℃下搅拌5~6小时,再降温至0~5℃搅拌析晶5~6小时,过滤,滤液套用于下批精制工艺,滤饼洗涤,干燥,得酯化物:16a-甲基-表氢化可的松-21丙酸酯106.8g,hplc含量97.2%,重量收率106.8%;
b、氧化物的制备:在一个1000ml三口瓶中,加入100g上述a所制备的酯化物,600ml氯仿,搅拌使之全溶,然后控温20~25℃下,慢慢滴加40g氧化铬与80g纯水配成的氧化剂溶液,约2.5~3小时内滴完,滴完后再保温于20~25℃继续反应1~2小时,tlc确认反应终点,反应完后,加入10g的保险粉以除去过量的氧化剂,然后减压浓缩回收约90%的氯仿,再降温至20~30℃,加入500ml纯水,于0~5℃搅拌析晶3~4小时,过滤,滤液排入废水处理池,滤饼洗涤干燥,得氧化物98.2g,hplc含量97.5%,重量收率98.2%;
c、双酯物的制备:在一个1000ml三口瓶中,加入100g上述b所制备的氧化物,300ml甲苯,40g丙酸酐,12g磷酸,保温于50~55℃下反应6~8小时,tlc确认反应终点,反应完后,降温至25~30℃,向反应液中慢慢滴加至80ml摩尔浓度为30%氢氧化钠溶液,分去水层,再用50ml纯水洗涤一次,分尽水,然后减压浓缩回收90~95%的溶剂甲苯,冷却后加入500ml纯水,于0~5℃搅拌析晶4~5小时,过滤,滤液排入废水处理池,滤饼水洗,干燥,得双酯物:16a-甲基-17a,21-双丙酰氧基-孕甾-4-烯-3,7,11,20-四酮111.8g,hplc含量98.2%,重量收率111.8%;
d、醚化物的制备:在一个1000ml三口瓶中,加入100g上述c所制备的双酯物,600ml乙醇,90ml原甲酸三乙酯和10g三氟乙酸,保温于20~25℃反应3~4小时,tlc确认反应终点,反应完后,加入适量的吡啶固体中和至溶液的ph6.5~7.5,减压浓缩回收90~95%的乙醇后,冷却降温至20~25℃,加入500ml纯水,于0~5℃搅拌析晶1.5~2.5小时,过滤,滤液排入废水处理池,滤饼洗涤干燥,得醚化物:3-乙烯醇醚基-16a-甲基-17a,21-双丙酰氧基-孕甾-3,5-二烯-7,11,20-三酮102.6g,hplc含量96.8%,重量收率102.6%;
e、还原物的制备:在一个2000ml三口瓶中,加入100g上述d所制备的醚化物,1200mlthf,保温于25~30℃下,搅拌使固体全部溶解,再慢慢加入18g硼氢化钾还原剂,约1.5~2.5小时加完,然后再于25~30℃继续保温反应1~2小时,tlc确认反应终点,反应完后,慢慢滴加100g摩尔浓度为50%的硫酸水溶液,使体系ph至2~3,再于25~30℃继续水解反应2~3小时,tlc确认反应终点,反应完后,慢慢加适量碱中和至体系ph至6~7,然后减压浓缩回收90~95%有机溶剂thf,再降温,慢慢加入600ml纯水水析,过滤,滤液排入废水处理池,滤饼洗涤干燥,得还原物粗品,将该粗品用500ml摩尔浓度为50%的酒精水溶液重结晶,得还原物:16a-甲基-7,11-二羟基-17a,21-双丙酰氧基-孕甾-4-烯-3,20-二酮70.8g,hplc含量97.6%,重量收率70.8%;
f、1-脱氢物的制备:在一个2000ml三口瓶中,加入100g上述e所制备的还原物,1200ml乙酸乙酯,85gddq脱氢剂,搅拌使其完全溶解,升温至75~80℃保温反应6~10小时,tlc确认反应终点,反应完后,滤除反应生成的氢醌,再加入适量的亚硫酸氢钠溶液破坏过量的ddq,然后减压浓缩回收90~95%有机溶剂乙酸乙酯,再降温,慢慢加入600ml纯水水析,离心,滤液排入废水处理池,滤饼洗涤干燥,得1-脱氢物粗品,将该粗品用500ml含20%酒精10%烧碱的水溶液重结晶,得1-脱氢物:16a-甲基-7,11-二羟基-17a,21-双丙酰氧基-孕甾-1,4-二烯-3,20-二酮62.4g,hplc含量97.2%,重量收率62.4%;
g、双丙酸阿氯米松的制备:在一个2000ml三口瓶中,加入100g上述f所制备的1-脱氢物,800ml甲苯,200g吡啶,升温搅拌使固体全部溶解后,再加入80g氯化亚砜氯化剂,保温于40~45℃搅拌反应4~6小时,tlc确认反应终点,反应完后,加入500g摩尔浓度为30%的烧碱溶液分2次中和洗涤至水溶液层ph6~7,减压浓缩回收90~95%的有机溶剂甲苯后,冷却降温至20~25℃,加入600ml纯水,于0~5℃搅拌析晶1.5~2.5小时,过滤,滤液排入废水处理池,滤饼洗涤干燥,得双丙酸阿氯米松粗品,将该粗品按实施例1中g所述方法重结晶,得双丙酸阿氯米松:7a-氯-16a-甲基-11-羟基-孕甾-1,4-二烯-3,20-二酮-17,21-双丙酸酯62.8g,熔点:214~215.5℃,hplc含量99.2%,重量收率62.8%。
以上内容是结合具体的优选实施方式对本发明作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明。对于本发明所属技术领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单推演和替换,都应当视为属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!