一种利用氧化还原电位调控吡啶衍生物催化加氢反应的方法与流程
本发明涉及一种吡啶衍生物催化加氢反应,具体地说是一种利用氧化还原电位调控吡啶衍生物催化加氢反应的方法,属于医药中间体制备领域。
背景技术:
哌啶、哌啶醇及其衍生物是一类重要的医药中间体,其广泛应用于糖尿病药物、抗抑郁药物、抗肿瘤药物、局麻药物以及降压药物合成中,其中,3-羟基哌啶主要用于合成依鲁替尼关键中间体(s)-1-叔丁氧羰基-3-羟基哌啶合成,4-羟基哌啶主要用于合成n-boc-4-羟基哌啶等,哌啶类衍生物在医药行业具有广泛用途和广阔市场前景。
目前,制备哌啶类衍生物最为常见的方法之一是直接对吡啶类衍生物进行催化加氢反应。催化加氢常用金属催化剂包括镍系(raneyni)、铑基、钌基、钯基等,对于上述反应,镍的催化活性较低,一般需200℃以上高温、10mpa以上高压反应,对生产设备要求较高;铑基催化剂效果较好,如:浙江司太立制药股份有限公司采用3-羟基吡啶经5%铑炭(rh/c)催化加氢制得消旋3-羟基哌啶,但是铑的价格相对较高(是钌的5~8倍左右),其生产成本很大,且污染相对较重。钌炭(ru/c)主要用于苯环还原及吡啶环还原,且以其流程少,转化率高,产率高,三废少等优点,引起了国内外极大关注。吡啶类衍生物催化加氢方面的代表性研究文献如下:
1、对于α-苯基-4-吡啶甲醇催化加氢反应,bela等以钯炭(pd/c)为催化剂、醋酸为反应介质,在60-80℃、8-10bar条件下,其反应底物在除去羟基同时吡啶环也被还原为哌啶环(europeanjournaloforganicchemistry2004,3623-3632)。
2、对于吡啶及其甲基吡啶衍生物催化加氢反应,ru/c催化活性高于pd/c、pt/c和ir/c(均为5%);在100℃、3.0mpa、ru/c与吡啶摩尔比=2.5/1000、乙醇中反应1h,其吡啶转化率大于99.9%,生成哌啶选择性为100%(催化学报,2006,27(10):921-926)。
3、对于3,5-二甲基吡啶催化加氢反应,周川利用单金属催化剂iii在130℃、9.5mpa、反应24h可得到82%产率,其中顺式-3,5-二甲基哌啶产物占比91%(3,5-二甲基吡啶催化加氢反应的研究[d].华中科技大学,2008)。
4、对于3-羟基吡啶催化加氢反应,凌洋以5%rh/c或pd/c为催化剂、纯水为反应溶剂,当rh/c添加量为底物质量5%,在80℃、3.0mpa反应2h,底物即可完全转化为3-羟基哌啶;若使用pd/c,其添加量10%,在100℃、7mpa下则需要反应38小时(手性3-羟基哌啶的合成[d].东南大学,2016)。
5、申请号200610088375.3,名称为“顺反式混合异构体3,5-二甲基哌啶的制备方法”,公开了一种由3,5-二甲基吡啶通过一步氢化制备顺式为(85±3)%、反式(15±3)%的3,5-二甲基哌啶混合物方法,该方法使用新型镍钌铑炭复合催化剂,以四氢呋喃为反应溶剂。
6、申请号200610144359.1,名称为“一种催化氢化酰基吡啶为取代哌啶化合物盐酸盐的方法”,公开了一种2-酰基吡啶或4-酰基吡啶在pd/c作用下催化氢化反应,其在反应体系中加入偕二氯烷烃,从而使该反应可在温和条件下顺利得到取代哌啶化合物盐酸盐。
7、申请号201510804425.2,名称为“一种制备3-羟基哌啶的方法”,公开了一种利用金属助剂镍提高铑碳对3-羟基吡啶催化氢化活性,从而使其在水或有机溶剂中、较低氢气压力下实现转化。
8、申请号201510771570.5,名称为“一种(s)-n-boc-3-羟基哌啶的合成方法”,公开了3-羟基吡啶在rh/c作用下于4-6mpa氢压、80-100℃反应32-60h,减压蒸馏得到3-羟基哌啶。
9、申请号201510178399.7,名称为“一种3-羟基吡啶液相催化加氢制备3-羟基哌啶的方法”,公开了3-羟基吡啶制备3-羟基哌啶的方法,该方法以ru/c、纯水为反应介质、反应温度140℃、氢压4.0mpa。
10、申请号201711448984.x,名称为“(s)-3-羟基哌啶的制备方法”,公开了钌/二氧化硅与三氧化二铝作为3-羟基吡啶催化加氢制备3-羟基哌啶的复配催化剂,该复配催化剂价格相对较低,但催化效果同样较好,且环境污染相对较小。
综上所述,目前对吡啶类衍生物催化加氢反应进程调控主要从催化剂(rh/c、ru/c、pd/c及复配ni-rh/c、ni-rh-ru/c)、反应温度、氢气压力及反应时间等方面开展,现有研究也表明催化剂活性、氢气压力、反应温度增加均可使反应进程加速。吡啶衍生物加氢反应本质是氧化还原反应,其氧化还原电位(orp)也是决定反应方向和程度的重要因素之一,这也使得通过orp调控吡啶衍生物催化氢化反应成为可能。理论上来说,反应起始物(或底物)orp会随介质ph变化而改变,吡啶衍生物所在介质体系的orp升高,其氧化性能也会有所提升,因此,有必要研究吡啶衍生物在不同介质体系的orp与底物转化、哌啶类产物含量之间的关联,以期实现利用orp调控吡啶衍生物催化加氢反应的目的。
技术实现要素:
本发明的目的在于提供一种利用氧化还原电位调控吡啶衍生物催化加氢反应的方法,以吡啶衍生物反应介质orp及其底物转化速度、哌啶产物含量数据分析为基础,从而筛选出哌啶衍生物较优的制备工艺。本发明工艺条件较为温和,且哌啶类产品具有纯度高、后处理方便等特点。
本发明以吡啶衍生物反应介质orp及其底物转化速度、哌啶产物含量数据分析为基础,从而筛选出哌啶衍生物较优的制备工艺,主要从以下几点进行优化筛选:
(1)将吡啶衍生物溶于水、不同浓度酸水溶液,获得底物进行催化加氢反应可能的介质环境,并检测其氧化还原电位(orp);以orp数值较高的介质进行催化加氢反应;
(2)以ru/c为催化剂,设计不同催化剂用量、氢气压力和反应温度方案进行加氢反应,定期监测各方案下底物转化速度、哌啶类产物含量与反应体系orp变化,初步建立加氢反应进程与起始介质orp之间的关联,并从中筛选出吡啶衍生物加氢反应介质较适orp范围、催化剂用量、反应温度及氢气压力;
(3)固定ru/c用量、反应温度和压力等条件,通过改变吡啶衍生物的底物浓度、介质酸度等,调整其orp在上述范围内并再次进行催化加氢反应,验证介质orp对反应进程的调控,进而确立哌啶衍生物较优制备工艺。
本发明筛选思路如下:(1)先根据反应实际设定酸水溶液浓度5-50%,而后初步固定底物浓度为1mol/l(这一浓度是氢化反应进行的常用浓度,而且底物浓度对orp影响不大,酸度影响更明显),进一步根据orp值筛选了酸浓度为30-50%的醋酸溶液、质量浓度5-15%的磷酸溶液或质量浓度5-15%的盐酸溶液;(2)结合反应进程监控,筛选了orp范围300-600mv以及氢化温度60-100度、压力2-5mpa;(3)通过底物浓度、酸度调节使orp在上述范围内,再结合反应进程调控,获得底物浓度在1-2.5mol/l反应速度都不错(浓度增大对反应进程加快并不明显),以1.5-2.0mol/l最优(主要考虑对于固定容积的氢化反应器来说,其投料量增大,生产效率也就提高了,但过大又导致搅拌受阻,故限定1.5-2.0mol/l;orp300-420mv。)
通过上述过程筛选优化后,本发明利用氧化还原电位调控吡啶衍生物催化加氢反应的方法,包括如下步骤:将反应底物吡啶衍生物溶于酸水溶液中,在催化剂的存在下进行催化加氢反应。
所述吡啶衍生物通式为
所述酸水溶液为醋酸溶液、盐酸溶液或磷酸溶液等;酸水溶液的质量浓度为5-50%。
进一步地,反应体系中吡啶衍生物浓度为1-2.5mol/l,优选1.5-2.0mol/l。
进一步地,所述酸水溶液优选为质量浓度30-50%的醋酸溶液、质量浓度5-15%的磷酸溶液或质量浓度5-15%的盐酸溶液。
所述催化剂为5%ru/c,催化剂的添加质量为吡啶衍生物质量的5-15%。
催化加氢反应过程中,氢气压力为2-5mpa,反应温度为60-100℃。
反应体系的orp为300-600mv,优选300-420mv。
本发明的优点及积极效果如下:
1、本发明借助吡啶衍生物在氢化反应体系相关orp数据与底物转化、哌啶类产物含量之间的关联筛选出哌啶衍生物较优制备工艺,这与传统的催化加氢反应筛选需要多轮试验相比,其筛选周期缩短、筛选成本降低。本发明为实现这一效果,orp数据和哌啶类产物含量分析是分步和有针对性地进行。第一步,对吡啶衍生物催化氢化反应的可能反应介质进行orp测定,分析底物在何种介质中orp增大(氧化性提高),以此结果筛选出吡啶衍生物加氢反应的较好介质;第二步,通过设计不同反应方案,并对不同方案下的底物转化、哌啶产物含量及orp数据进行分析,初步建立orp与反应进程的关联,以此选择吡啶衍生物加氢反应介质较合适的orp范围,通过该步骤也可判定第一步中根据orp数据初选的介质体系是否符合预期;第三步固定加氢反应中其它因素(如:催化剂种类、用量、氢气压力、反应温度),通过底物浓度及介质ph调整orp在上述选定范围内,再次通过实验确认哌啶产物较优制备工艺,这种基于orp优化制备工艺的思路缩短了试验周期、降低了实验消耗。
2、本发明提出基于orp分析指导吡啶衍生物催化氢化反应的方法,扩大了orp应用范围。传统的orp调控用以水质监测、金属腐蚀、资源勘测、土壤环境监测等领域,也有将orp控制策略应用于琥珀酸、柠檬酸、乳酸、木糖醇、1,3-丙二醇、乙醇等生物发酵过程优化(申请号200810034199.4;bioprocessandbiosystemsengineering,2010,33(8):911-920;化工学报),2009,60(10):2555-2561;foodtechnologyandbiotechnology,2000,38(3):193-201;appliedbiochemistryandmicrobiology,2011,47(1):33-38;appliedmicrobiologyandbiotechnology,2003,63(1):96-100;appliedmicrobiologyandbiotechnology,2006,69:554-563;化工学报,2012,63(4):1168-1174journalofbiotechnology,2011,153(1):42-47)。
3、本发明利用反应体系orp与底物转化速度、哌啶产物含量之间的关联筛选出哌啶衍生物较优制备方法,其产物纯度高(≥98%,如附图2中4-羟基哌啶盐酸盐)、后处理方便。与纯水中进行的催化加氢反应相比,该工艺降低了氢气压力和反应温度,同时所得的哌啶盐产品吸湿性有所改善(如:4-羟基哌啶约500mg置于空气中放置1h,其质量增加4.0~4.5%;而4-羟基哌啶醋酸盐放置1h质量增加不足1.5%;4-羟基哌啶盐酸盐置于空气中放置1h质量增加约2.5-3.0%)。此外,也可在反应结束后进行碱化处理,再通过减压蒸馏也可方便获得游离的哌啶类产物。
附图说明
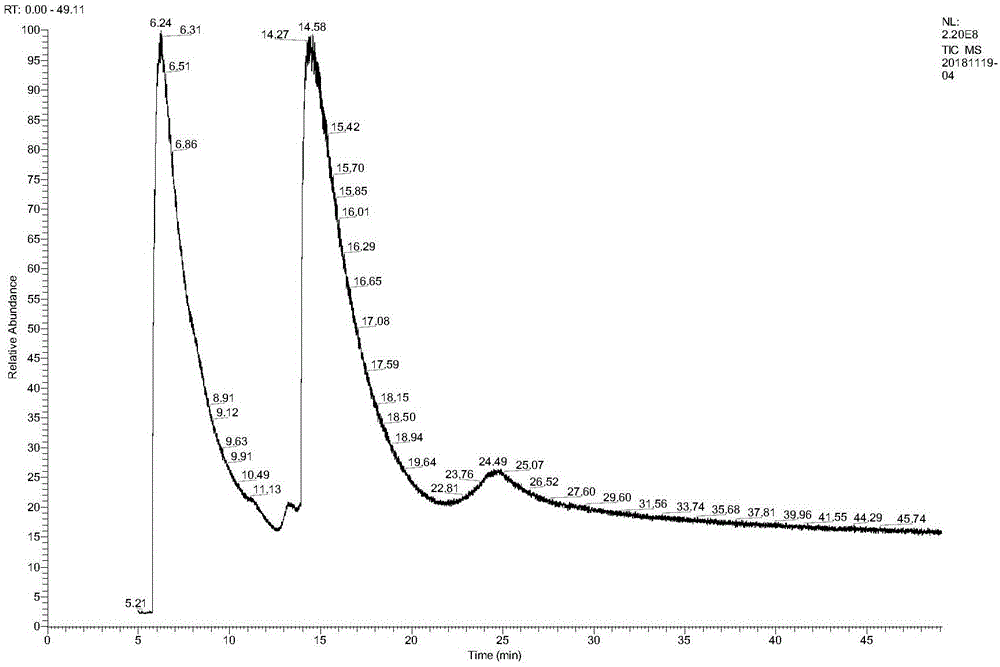
图1是4-羟基吡啶催化反应体系物质组成gc-ms总离子流图。从图中可以看出,4-羟基哌啶(rt=6.24min)及4-羟基吡啶(rt=14.27min)可完全分离,反应进程中4-羟基吡啶的转化以及4-羟基哌啶含量可借助gc面积归一化法进行监测。
图2是4-羟基吡啶在盐酸体系下催化氢化所得产物的核磁共振谱图。从图可以看出,4-羟基哌啶盐酸盐纯度已很高(>98%),这也说明orp较高的酸性介质体系有利于催化加氢反应,且反应体系只需经过简单减压浓缩即可获得高纯度4-羟基哌啶盐酸盐产物。
具体实施方式
下面通过具体实施例对本发明作进一步详述,以下实施例只是描述性的,不是限定性的,不能以此限定本发明的保护范围。
实施例1:
本实施例中借助orp数据筛选4-羟基吡啶催化加氢反应的方法如下:
1、将1.9g4-羟基吡啶溶于15-20ml水、30%-50%冰醋酸及5-15%盐酸水溶液(v/v),获得底物进行催化加氢反应的可能介质环境;
2、对上述各体系orp分别进行检测,基于测定结果有针对地选择4-羟基吡啶可能具有较高氧化性能的25%醋酸、40%醋酸、5%盐酸及10%盐酸水溶液进行催化加氢反应;
3、将19g4-羟基吡啶溶于约200ml醋酸或盐酸水溶液,以5%ru/c催化、添加量为5%、10%、15%底物质量;反应釜中氢气压力控制在3-5mpa、温度80-100℃,待氢气压力不再降低时分别取样监测,对不同方案下反应液过滤,进行orp及4-羟基哌啶产物的gc或gc-ms(附图1)测定,结合4-羟基吡啶转化、4-羟基哌啶含量及起始orp等结果分析4-羟基吡啶加氢反应较合适反应介质orp范围为320-400mv、催化剂添加量5~10%;氢气压力3.5mpa,温度95-100℃;
4、固定ru/c添加量8%、反应压力3.5mpa及温度95℃,调整4-羟基吡啶底物浓度1.5-2.0mol/l及介质酸度,使其orp在330-350、370-390mv附近,然后再次进行催化加氢反应,反应分别在16h、24h取样,gc面积归一化法检测不同orp方案下4-羟基吡啶与4-羟基哌啶面积百分比分别为:
综合考虑4-羟基吡啶转化速度及4-羟基哌啶产物含量等数据,优选出4-羟基哌啶较优制备工艺为:2mol/l底物浓度、35%醋酸水溶液(orp340mv左右)或10%盐酸水溶液(orp380mv左右)为反应介质、5%钌炭催化添加量8%、温度95℃、氢气压力3.5mpa,在此条件下完成反应约需24h。
实施例2:
本实施例中借助orp数据筛选3-羟基吡啶衍生物催化氢化反应的方法如下:
1、将1.9g3-羟基吡啶溶于15-20ml水、30%-50%冰醋酸及5-10%磷酸水溶液(v/v),获得底物进行催化加氢反应的可能介质体系;
2、对上述各体系orp分别进行检测;基于测定结果有针对地选择3-羟基吡啶可能具有较高氧化性能的35%醋酸、50%醋酸及10%磷酸水溶液进行催化加氢反应;
3、将19g3-羟基吡啶溶于约200ml醋酸或磷酸水溶液,以5%ru/c催化、添加量为10%、15%底物质量;反应釜中氢气压力控制在3-5mpa、温度70-100℃,待氢气压力不再降低时分别取样监测。对不同反应阶段反应液过滤,进行orp及哌啶含量测定,结合底物转化、哌啶含量及起始orp变化等结果分析3-羟基吡啶加氢反应较合适的反应介质orp范围为350-400mv、催化剂添加量10%、氢气压力4mpa,温度100℃;
4、固定ru/c催化剂添加量10%、反应压力4mpa及温度100℃,调整3-羟基吡啶体系浓度1.5-2.0mol/l及酸水反应介质酸度,使其orp在360-380、390-400mv附近,然后再次进行催化加氢反应,反应分别在15h、20h取样,gc面积归一化法检测不同orp方案下3-羟基吡啶与3-羟基哌啶面积百分比分别为:
综合考虑3-羟基吡啶转化速度及3-羟基哌啶产物含量等数据,优选出3-羟基哌啶较适制备工艺为:1.5-2.0mol/l底物浓度、40%醋酸(orp360mv)水溶液为反应介质、5%ru/c催化添加量10%、温度100℃、氢气压力4mpa,在此条件下完成反应约需20h。
实施例3:
本实施例中借助orp数据筛选2-甲基吡啶衍生物催化氢化反应的方法如下:
1、将1.88g2-甲基吡啶溶于15-20ml水、30%-50%冰醋酸及5-15%盐酸水溶液(v/v),获得底物进行催化加氢反应的可能介质环境;
2、对上述各体系orp分别进行检测;基于测定结果有针对地选择2-甲基吡啶可能具有较高氧化性能的35%醋酸、5%盐酸及10%盐酸水溶液进行催化加氢反应;
3、将18.8g2-甲基吡啶溶于约200ml醋酸或盐酸水溶液,以5%ru/c催化、添加量为5%、10%底物质量;反应釜中氢气压力控制在2-4mpa、温度80-100℃,待氢气压力不再降低时,对不同方案下反应液过滤,进行orp及2-甲基哌啶含量测定。结合2-甲基吡啶转化、2-甲基哌啶含量及起始orp变化等结果分析2-甲基吡啶催化加氢反应介质较合适orp范围为360-385mv、催化剂添加量5-10%、反应压力2.5mpa及温度90℃;
4、固定ru/c添加量8%、反应压力2.5mpa及温度90℃,调整2-甲基吡啶的反应浓度2mol/l及酸水反应介质酸度,使其orp在370-375mv附近,然后再次进行催化加氢反应,反应约4h、6h取样,gc面积归一化法检测不同orp方案下3-羟基吡啶与3-羟基哌啶面积百分比分别为:
结合2-甲基吡啶转化速度及2-甲基哌啶产物含量等数据,优选2-甲基哌啶较适制备工艺为:2mol/l底物浓度、7-9%hcl水溶液(orp370-380mv左右)为反应介质、5%ru/c添加量8%、温度90℃、氢气压力2.5mpa,在此条件下完成反应约需6h。
实施例4:
本实施例中借助orp数据筛选3,5-二甲基吡啶衍生物催化氢化反应的方法如下:
1、将2.14g3,5-甲基吡啶溶于15-20ml水、30%-50%冰醋酸、5-15%盐酸水溶液(v/v)、5-10%磷酸水溶液(v/v),获得底物进行催化加氢反应的可能介质体系;
2、对上述各体系orp分别进行检测;基于测定结果有针对地选择3,5-甲基吡啶可能具有较高氧化性能的40%醋酸、10%盐酸水溶液进行催化加氢反应;
3、将21.4g3,5-二甲基吡啶溶于约200ml醋酸或盐酸水溶液,以5%ru/c催化、添加量为5%、10%底物质量;反应釜中氢气压力控制在3-4mpa、温度80-100℃,待氢气压力不再降低时分别取样监测3,5-二甲基吡啶转化情况,对不同反应阶段反应液过滤,进行orp及哌啶含量测定,结合原料转化、哌啶含量及起始orp变化等结果分析3,5-二甲基吡啶加氢反应较合适的反应介质orp范围为370-410mv、催化添加量10%、反应压力3.0mpa及温度90℃;
4、固定5%ru/c催化添加量10%、反应压力3.0mpa及温度90℃,调整3,5-二甲基吡啶体系浓度1.5mol/l及酸水反应介质酸度,使其orp在375-385、395-405mv附近,然后再次进行催化加氢反应,反应约6h、8h取样,gc面积归一化法检测不同orp方案下3,5-二甲基吡啶与3,5-二甲基哌啶面积百分比分别为:
综合考虑3,5-甲基吡啶转化速度及3,5-二甲基哌啶产物含量等数据,优选出3,5-二甲基吡啶较适制备工艺为:1.5mol/l底物浓度、10%hcl(orp380mv)或45%醋酸水溶液(orp380mv)为反应介质、5%ru/c催化添加量10%、温度90℃、氢气压力3.0mpa,在此条件下完成反应约需8h。
- 还没有人留言评论。精彩留言会获得点赞!