一种肠道菌群16SrRNA基因的检测方法及其应用与流程
本发明属于微生物和基因工程技术领域,具体涉及一种肠道菌群16srrna基因的检测方法,以及在肝病肠道菌群检测、肝病风险预测中的应用。
背景技术:
在人类的肠道中,存在着大量微生物。这些肠道微生物除了传统意义上的肠道细菌群,还包括古细菌、病毒和原生动物,其中98%以上是细菌,统称为肠道菌群。正常肠道菌群有500~1500种不同细菌种类,其中绝大多数为厌氧菌。从基因层面上看,人体自身的基因组大约携带了2.5万个基因,而人体肠道微生物编码的基因总数约是人自身基因总数的150倍,被视作人类“第二基因组”。人体肠道微生物的全部基因信息称为人体肠道宏基因组(matagenome)。
肠道既是吸收营养物质的主要器官,也是机体抵御食物中有害物质的重要免疫屏障,更与肝脏的免疫细胞构成免疫平衡,共同维持机体稳态。研究表明,肝病的发生、发展与肠道菌群的变化关系紧密,肠道菌群与肝脏通过门静脉在解剖及功能上有着密切联系,它们的病理生理学联系被称为“肠-肝轴”。肝脏的门静脉是肠道各种抗原、毒素、肠道菌群及其产物侵入的第一道防线,这种关系赋予了肝脏重要的代谢、免疫及解毒功能。肠道、肝脏及免疫系统的相互作用是确保肝脏健康的重要因素之一。
慢性肝病时,20%~75%的患者会出现肠道菌群失调,且菌群失调的严重程度和肝病严重程度相关。从肝炎、肝硬化到肝癌,肝病的发展过程可促使肠道菌群紊乱及肠道黏膜屏障功能障碍的发展。但是目前对这种菌群失衡影响的认识仍很局限。随着技术手段的进步,研究肠道微生物的方法已经从早期的细菌分离培养和基于16s基因的聚合酶链式反应-变性梯度凝胶电泳、限制性片段长度多态性、16s芯片及16s克隆文库测序等,逐渐发展到16s高通量测序和宏基因组测序。随着高通量测序技术的发展,从宏基因组水平研究患者肠道菌群的总体构成特点,有助于更加全面的理解菌群对宿主生理和病理的影响,为疾病的预防、诊断和治疗提供参考依据,进而为临床提供更为准确有效的益生菌制剂,延缓病情进展及改善预后。
技术实现要素:
本发明的目的在于提供一种肠道菌群16srrna基因的简便的检测方法,并结合二代测序技术和生物信息学分析方法,提出一种能够有效检测分析肝病患者肠道菌群的方法,为肝病的辅助诊断与治疗方案选择提供参考。
本发明技术方案如下:
一种肠道菌群16srrna基因的检测方法,所述的检测方法包括粪便样本的裂解,肠道菌群16srrna基因文库的构建,16srrna基因文库的测序,测序数据的分析。
所述的粪便样本的裂解方法包括:将样本与裂解液混合,70-95℃孵育10-20min,涡旋混合后离心,取上清即可。
其中,所述的裂解液的成分包括0.1-0.2mol/ltris-hcl,0.05-0.1mol/ledta,0.5-1.0mol/lnacl,2%-4%聚乙烯吡咯烷酮,1%-2%sds,ph7.0-8.0。
所述的肠道菌群16srrna基因文库的构建方法包括:将样本裂解后获得的上清液,特异性引物进行pcr扩增和靶点标记;pcr产物纯化;为靶点建立index标签获得文库;文库纯化;文库质检,质检合格后即可上机测序。
其中,所述的特异性引物的序列如下:
上游引物f:
tcgtcggcagcgtcagatgtgtataagagacagactcctacgggaggcagcag(seqidno:1)
下游引物r:
gtctcgtgggctcggagatgtgtataagagacagggactachvgggtwtctaat(seqidno:2)
所述的测序数据的分析方法包括:对文库测序后得到的数据进行拼接,过滤除去引物和嵌合体序列,进行otu聚类和物种注释,得到肠道菌群样本中的物种多样性和物种丰度,将其与健康人的样本进行比较即可。
进一步的,所述的肠道菌群16srrna基因的检测方法在肝病患者肠道菌群检测中的应用。
进一步的,所述的肠道菌群16srrna基因的检测方法在肝病患病风险预测中的应用。其中,所述的应用方法包括:先对肠道菌群16srrna基因进行测序;再根据测序数据进行物种注释和物种多样性分析;然后根据回归模型y=α+β1×x1+β2×x2+…+β15×x15,式中α为常数,β为回归系数,x为致病菌属的相对丰度,y为预测值,得出风险预测值;根据风险预测值判断患肝病的风险大小。
区别于现有技术,上述技术方案的有益效果如下:
(1)本发明采用粪便直接裂解的方法获得肠道菌群的dna,比传统的dna提取方法如磁珠法或离心柱法,更简便、快速,并且构建的文库质量较好,与传统dna提取方法得到的dna构建的文库质量基本一致。
(2)本发明可用于肝病的辅助诊断,为疾病的预防、诊断和治疗提供参考依据。
附图说明
图1为具体实施方式所述的pcr扩增后的dna电泳图,(a)图为第一轮pcr扩增的dna电泳图,(b)图为第二轮pcr扩增后的dna电泳图,(a)、(b)图中靠左侧的第一条泳道为markerdl2000,条带大小从上到下依次为:2000、1000、750、500、250、100bp。
图2为具体实施方式所述的文库质检结果。
图3为具体实施方式所述的测序数据质量结果。
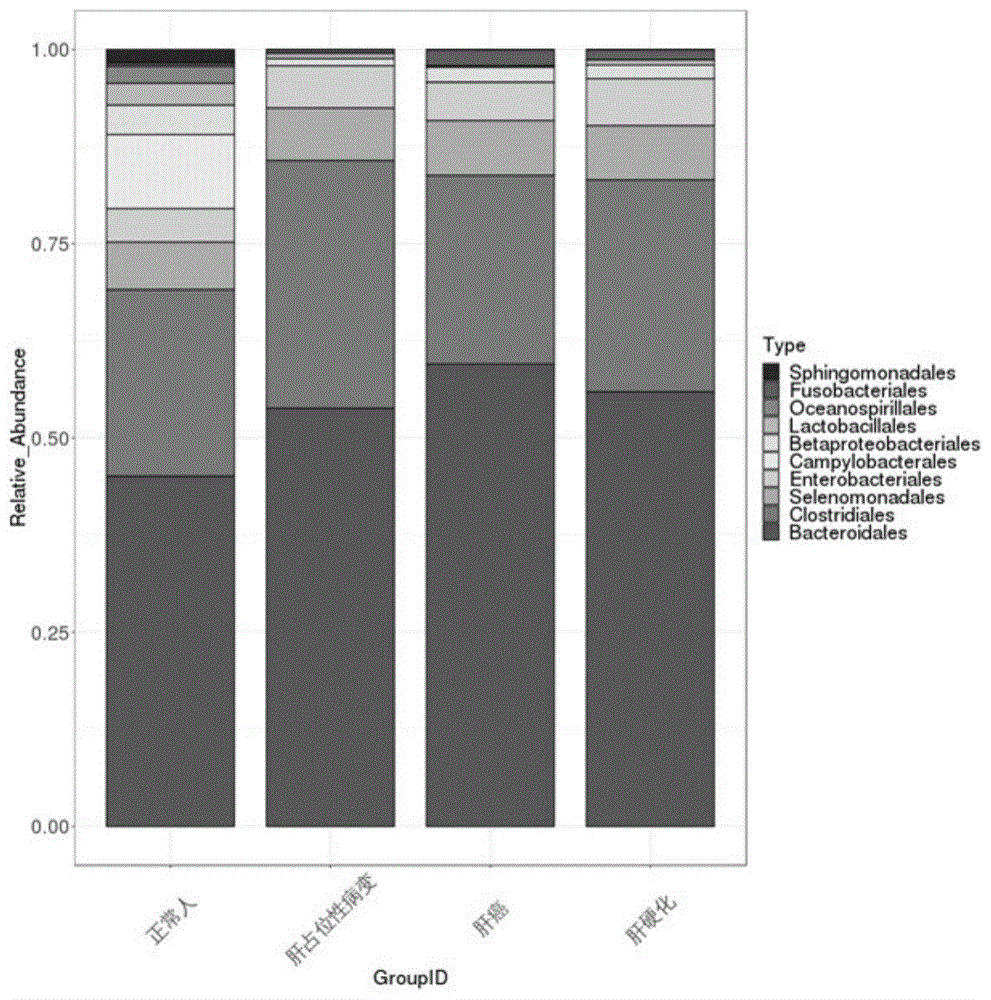
图4为具体实施方式所述的物种相对丰度柱形累加图。
图5为具体实施方式所述的auc曲线。
具体实施方式
下面结合具体实施例,对本发明的技术方案进行详细说明,需要说明的是,这些实施例仅仅是说明性的,而不能理解为对本发明的限制。
若未特别指明,实施例中所采用的技术手段为本领域技术人员所熟知的常规手段,可以参照《分子克隆实验指南》第三版或者相关产品进行,所采用的试剂和产品也均为可商业获得的。未详细描述的各种过程和方法是本领域中公知的常规方法,所用试剂的来源、商品名以及有必要列出其组成成分者,均在首次出现时标明,其后所用相同试剂如无特殊说明,均以首次标明的内容相同。
实施例1肠道菌群16srrna基因的建库
1样本来源
样本在福建地区收集,总共收集健康人的新鲜粪便样本101例、肝硬化患者的新鲜粪便样本112例、肝占位性病变患者的的新鲜粪便样本41例、肝癌患者的新鲜粪便样本20例,-80℃冻存。
2粪便样本的裂解方法
2.1样本裂解液的配制
裂解液glb:0.1-0.2mol/ltris-hcl,0.05-0.1mol/ledta,0.5-1.0mol/lnacl,2%-4%聚乙烯吡咯烷酮(pvp),1%-2%sds,ph7.0-8.0。
2.2步骤
a)向样本中加入1.2ml裂解液glb,间歇振荡1-2min至样本混匀。
b)70℃孵育10-20min。
注意:对于较难破壁的革兰氏阳性菌,可将温度提高至95℃以促进裂解。
c)涡旋15-30sec,12,000rpm(~13,400×g)离心10min。转移上清液700μl至新的2ml离心管。
3文库构建
3.1引物设计
针对16srrna基因的v3-v4区设计特异性引物并加上overhang序列,具体序列如下:
上游引物f:
tcgtcggcagcgtcagatgtgtataagagacagactcctacgggaggcagcag(seqidno:1)
下游引物r:
gtctcgtgggctcggagatgtgtataagagacagggactachvgggtwtctaat(seqidno:2)
3.2pcr扩增和标记靶点
a)从-20℃冰箱中取出pcramplificationmix(购自南京诺唯赞生物科技有限公司),primermix1(含有上述引物的tebuffer)和肠道菌群基因组dna,并在冰上进行化冻;
b)在0.2mlpcrtube中配制反应体系
c)将配置好的mixure轻柔吹打10次,混匀,置于pcr仪中,执行以下程序:
d)反应结束后,取2μlpcr产物进行琼脂糖凝胶电泳,pcr产物约在500bp。电泳条件:1%agarose,d2000ladder,120v,20min。
3.3pcr产物纯化
a)预先将ampurexp磁珠从4℃冰箱中取出,室温平衡30min,使用前振荡混匀。新鲜配置80%乙醇;
b)取新的1.5mlep管,标记后加入18.4μl磁珠,然后加入上一步的pcr产物,(r1=0.8:1)吹打10次混匀,注意不要产生气泡,室温静置5min;
c)离心后,将离心管置于磁力架上10min至液体澄清;
d)小心去除上清,注意尽量不要吸到磁珠;
e)保持ep管在磁力架上,贴壁缓慢加入200μl80%的乙醇,吸打后静置30sec,去上清;
f)重复洗涤一次,去上清时尽可能除去任何残留的乙醇;
g)保持ep管在磁力架上,室温静置4min干燥磁珠;
h)加入32μlresuspensionbuffer,用枪吹打混匀磁珠,然后室温静置2min。
i)瞬时离心后,将离心管放到磁力架上至液体澄清。
j)移取30μl上清至新的标记的1.5ml离心管中。
3.4为靶点建立index标签
a)从-20℃冰箱中取出pcramplificationmix(2x),10μm/μlindexedpcrprimeri5和indexedpcrprimeri7,并在冰上进行化冻;
以下仅列出一组index序列,其余序列见nexteraindexkit–pcrprimers试剂盒。
i5:aatgatacggcgaccaccgagatctacacctctctattcgtcggcagcgtc(seqidno:3)
i7:caagcagaagacggcatacgagattcgccttagtctcgtgggctcgg(seqidno:4)
b)在0.2ml离心管中按以下方法配制反应体系:
c)将配置好的mix轻柔吹打10次混匀,然后置于pcr仪中,反应程序如下:
3.5文库纯化
a)取新的1.5ml离心管,标记后加入50μl室温平衡后的磁珠,然后加入上一步的pcr产物,吹打10次混匀,室温静置5min;
b)瞬时离心后,将离心管放到磁力架上至液体澄清。
c)小心去除上清,注意尽量不要吸到磁珠。
d)保持pcr管在磁力架上,加入200μl80%的乙醇,吸打后静置30sec,去上清。
e)重复洗涤一次,去上清时尽可能除去任何残留的乙醇。
f)保持离心管在磁力架上,室温静置4min干燥磁珠。
g)加入27μlresuspensionbuffer,用枪吹打混匀磁珠,然后室温静置2min。
h)瞬时离心后,将离心管放到磁力架上至液体澄清。
i)移取25μl上清至新的标记的1.5ml离心管中。
3.6文库质检
a)取1μl使用qubit3.0fluorometer(qubitdsdnahsassaykit)进行浓度检测,并记录浓度。
b)使用agilent2200highsensitivityd1000kit对文库进行条带分布检测。
4实验结果
4.1dna起始量优化实验结果
dna建库起始量为12.5ng和25ng的实验体系摸索结果如表1所示,当dna起始量为12.5ng时,嵌合体(chimeras)产生较少,所以最终选择dna建库起始量为12.5ng。
表1dna建库不同起始量的实验结果
4.2pcr循环数优化实验结果
pcr循环数越高,会导致越多的嵌合体,导致有效数据降低,因此,本次实验设计3个不同循环数,进行实验体系摸索,实验结果如表2所示。实验结果显示,pcr循环数为25时,嵌合体产生较少,所以选择pcr循环数为25个循环。
表2不同pcr循环数的实验结果
4.3pcr扩增结果
第一轮和第二轮pcr结果扩增出的目标片段在500-750bp之间,见图1,图中120bp左右为引物二聚体。
4.4文库纯化及质检结果
文库经磁珠法纯化后,引物二聚体可被去除。构建好文库用agilent2200bioanalyzer进行质检,结果如图2所示,文库大小为500-700bp,基本上没有120bp左右的引物二聚体。
采用裂解方法得到的dna构建的文库质量与磁珠法或离心柱法提取得到的dna获得的文库质量基本一致,因此可以采用裂解法获得dna构建文库。
实施例2文库的测序
1测序步骤
1.1文库混合
将需要测序的文库,根据需要测的3万tags数要求混合;
1.2文库质检
将混合后的样本,进行qubit3.0和agilent2200bioanalyzer质检;
1.3文库稀释
用hybbuffer对文库进行稀释到上机浓度;
1.4文库变性
用0.2nnaoh对文库进行变性5分钟,变性后可选0.2m的tris-hclph7.0进行文库变性后中和;
1.5变性稀释
用hybbuffer对变性文库进行稀释;
1.6上机稀释
最后,再用hybbuffer对变性稀释文库进行最后上机稀释;
1.7上机测序
将上机稀释文库,放进试剂槽中,和芯片等一同放进miseq测序仪,导入samplesheet,设置reads和index的读长等,最后仪器开始测序。
2测序结果
比较了miseqpe250和miseqpe300的测序质量,实验结果显示,miseqpe300获得的有效tags在3万-5万条之间,q30在65%-70%之间;而miseqpe250获得的有效tags可达在3.5万-6万条之间,q30在70%-80%之间。说明较miseqpe300的测序质量好。
选用miseqpe250进行测序,整个run的结果,q30>75%,碱基质量较好,见图3。
实施例3测序数据的分析
1测序数据拼接
测序仪产生的图像数根据barcode序列从下机数据中拆分出各样品数据,截去barcode后使用flash对每个样品的reads进行拼接,得到的拼接序列为原始tags数据(rawtags);拼接得到的rawtags,需要经过严格的过滤处理得到高质量的tags数据(cleantags)。参照qiime的tags质量控制流程,进行如下操作:a)tags截取:将rawtags从连续低质量值(默认质量阈值为<=19)碱基数达到设定长度(默认长度值为3)的第一个低质量碱基位点截断;b)tags长度过滤:tags经过截取后得到的tags数据集,进一步过滤掉其中连续高质量碱基长度小于tags长度75%的tags。经过以上处理后得到的tags去除引物以及嵌合体。
2测序数据处理
使用mothur过滤处理得到高质量的reads,之后进一步去除嵌合体序列,至此完成测序数据的处理。
3otu聚类和物种注释
利用mothur软件对数据处理过的样品进行聚类,默认以97%的一致性(identity)将序列聚类成为otus(operationaltaxonomicunits),同时会选取otus的代表性序列,依据其算法原则,筛选的是otus中出现频数最高的序列作为otus的代表序列。对otus代表序列进行物种注释,用mothur方法与silva的ssurrna数据库进行物种注释分析(设定阈值为0.8~1),获得分类学信息并分别在各个分类水平:kingdom(界),phylum(门),class(纲),order(目),family(科),genus(属),species(种)统计各样本的群落组成。
根据物种注释结果,选取每个样品或分组在各分类水平上最大丰度排名前10的物种,生成物种相对丰度柱形累加图,以便直观查看各样品在不同分类水平上,相对丰度较高的物种及其比例。
4物种多样性及组间差异分析
根据物种注释结果,进行alpha多样性指数组间差异分析,并通过t-test检验分析组间物种多样性差异是否显著。
采用lefse(ldaeffectsize)方法,在组与组之间寻找具有统计学差异的biomarker,即组间差异显著的物种。lefse是一种用于发现高维生物标识和揭示基因组特征的软件,包括基因、代谢和分类,用于区别两个或两个以上生物条件(或者是类群)。该算法强调的是统计意义和生物相关性。lefse的统计结果包括三部分,分别是lda值分布柱状图,进化分支图(系统发育分布)和组间具有统计学差异的biomarker在不同组中丰度比较图。
5分析结果
根据物种注释结果,得到目水平上的物种相对丰度柱形累加图,如图4所示。与健康人相比,肝病患者肠道菌群多样性降低,肝硬化、肝占位性病变和肝癌患者肠道菌群中潜在致病菌增多、有益菌减少。其中,肝硬化和肝癌患者肠道菌群的多样性结构相似,肝占位性病变患者的肠道菌群多样性最低。
根据lefse分析结果,并结合t-test检验结果,分析得出普氏菌属(prevotella)、毛螺菌属(lachnospira)、萨特氏菌(sutterella)、考拉杆菌属(phascolarctobacterium)等15个属是肝病主要致病菌属,可以作为肝病相关的生物标志物,其中普氏菌属(prevotella)为最主要的致病菌。
实施例4诊断模型
根据实施例3分析得到的菌群数据和肝病生物标志物作逻辑回归分析。建立回归模型:y=α+β1×x1+β2×x2+…+β15×x15,式中α为常数,β为回归系数,x为致病菌属的相对丰度,y为预测值。
根据proc包画图找到最优临界点为0.337。也就是说,当预测值大于0.337被分类为正常人,预测值小于0.337被分类为肝病患者时,预测效果最好。计算得到精度(accurancy)为0.741,说明这个模型拟合效果不错。
通过画roc曲线,并计算其auc面积,作为评估二类分类效果的一个典型测量。计算后得到auc为0.733,如图5所示。说明本模型预测效果较好,可以根据该模型对肝病的患病风险进行预测。
综上,首先,物种多样性、相对丰度以及肝病相关致病菌属,可以作为肝病辅助诊断的指标。其次,还可以结合诊断模型,对肝病进行进一步诊断,或者对肝病患病风险进行分析。再次,肝病患者可以通过补充缺失的有益菌群,辅助肝病的治疗。
需要说明的是,尽管在本文中已经对上述各实施例进行了描述,但并非因此限制本发明的专利保护范围。因此,基于本发明的创新理念,对本文所述实施例进行的变更和修改,或利用本发明说明书及附图内容所作的等效结构或等效流程变换,直接或间接地将以上技术方案运用在其他相关的技术领域,均包括在本发明的专利保护范围之内。
序列表
<110>福建西陇生物技术有限公司,福州福瑞医学检验实验室有限公司
<120>一种肠道菌群16srrna基因的检测方法及其应用
<130>2019
<160>4
<170>siposequencelisting1.0
<210>1
<211>53
<212>dna
<213>细菌(bacteria)
<400>1
tcgtcggcagcgtcagatgtgtataagagacagactcctacgggaggcagcag53
<210>2
<211>54
<212>dna
<213>细菌(bacteria)
<400>2
gtctcgtgggctcggagatgtgtataagagacagggactachvgggtwtctaat54
<210>3
<211>51
<212>dna
<213>细菌(bacteria)
<400>3
aatgatacggcgaccaccgagatctacacctctctattcgtcggcagcgtc51
<210>4
<211>47
<212>dna
<213>细菌(bacteria)
<400>4
caagcagaagacggcatacgagattcgccttagtctcgtgggctcgg47
- 还没有人留言评论。精彩留言会获得点赞!