一种新型氮杂稠环化合物及其制备方法和应用与流程
本发明涉及有机半导体材料技术领域,更具体地,涉及一种新型氮杂稠环化合物及其制备方法和应用。
背景技术:
有机半导体材料与无机半导体材料相比,突出的优势包括四个方面:1)有机物可以通过简单的化学修饰进行改性,制得人们需要的材料,甚至可以得到多功能性器件;2)有机半导体材料具有良好的柔性和韧性,如果再利用聚合物等柔性衬底,就可以全柔性器件,进而将有机场效应晶体管应用于集成电路、柔性显示、电子纸张等可卷曲、可折叠产品中;3)通过简单的化学修饰,可以使有机半导体溶于常见的溶剂中,进而用溶液处理的方法代替传统的真空沉积方法来制备器件(喷墨打印、旋涂、滴注、微接触印刷等),将大大简化器件的处理工艺、节约成本,并且有利于制备大规模集成电路;4)有机半导体材料无论从材料的合成以及器件的制备等方面都具有非常大的降低成本的潜力。
而在有机小分子材料领域中,氮杂芳香环化合物在有机场效应晶体管应用中作为有机光电材料具有很大的潜在价值。
在氮杂场效应材料当中,酞菁类材料是研究比较热门的氮杂环材料,国际上的相关研究制得的器件也取得了较好的迁移率性能。而将氮原子应用到并苯体系当中来提高材料的稳定性,这方面的研究也比较多,缪谦课题组合成了一系列并五苯类似物,拥有较好的迁移率。
吲哚咔唑类衍生物也被广泛研究,吲哚咔唑类衍生物与上面所述的氮杂化合物的最大区别是,氮杂单元从六元环变成了五元环,较小环的张力使分子在进行一定的烷基等修饰时仍然能保持刚性,进而保持分子的平面结构。这对于提高材料的稳定性以及溶解度是大为有利的。
然而现有的材料和合成方法难以做到稠合有效苯环长度过长的分子,并且获得的材料性能和稳定性也不太理想。
技术实现要素:
本发明的目的在于克服现有的氮杂芳香环化合物存在的缺陷和不足,提供一种新型的氮杂稠环化合物的合成方法,得到有效共轭长度较长的大分子平面结构的氮杂稠环化合物,并且合成的材料拥有优异的迁移率性能,在有机光电领域拥有极其广泛的应用前景。
本发明的目的还在于提供所述新型的氮杂稠环化合物的制备方法。
本发明的目的还在于提供所述氮杂稠环化合物的应用。
本发明的上述目的是通过以下技术方案给予实现的:
一种新型氮杂稠环化合物,所述化合物的化学结构式通式如式(ix)、(x)、(xi)所示:
其中,ar为苯环个数≥0的稠环、苯环并氮杂环、稠环并氮杂环、苯环接取代基并氮杂环或稠环接取代基并氮杂环,r为碳原子数≥12的直链或支链烷基链。
优选地,所述化合物的化学结构式如式(iii)、式(vi)、式(d)、式(f)、式(g)、式(h)、式(i)、式(j)、式(k)所示:
其中,r为碳原子数≥12的直链或支链烷基,r’为氟、氯、溴三种卤代基中的一种。
优选地,所述r为十二烷基、2-辛基十二烷基等碳原子数≥12的直链或支链烷基。
本发明还保护上述任一项所述的氮杂稠环化合物的制备方法,包括如下步骤:
对于通式(ix)类,在氮气环境下,将2当量1,2-稠环或稠环并氮杂环或稠环(含卤代基)并氮杂环二胺(式vii),与1当量的3,8-双(2-r烷基链)-3,8-二氢吲哚[7,6]吲哚-1,2,6,7-四酮(式viii)加入到反应容器中,然后加入有机溶剂,加热至回流,70~120℃反应1~24h。将得到的溶液先减压蒸馏除去溶剂,再通过层析柱纯化,分离提纯之后减压蒸馏旋干洗脱剂即可得到产物(式ix);其化学反应方程式如下所示:
对于通式(x)类,在氮气环境下,将1当量1,2-稠环或稠环并氮杂环或稠环(含卤代基)并氮杂环二胺(式vii),与1当量的3,8-双(2-r烷基链)-3,8-二氢吲哚[7,6]吲哚-1,2,6,7-四酮(式viii)加入到反应容器中,然后加入有机溶剂,70~80℃反应2~4h。将得到的溶液先减压蒸馏除去溶剂,再通过层析柱纯化,分离提纯之后减压蒸馏旋干洗脱剂即可得到产物(式x);其化学反应方程式如下所示:
对于通式(xi)类,其步骤为:在氮气环境下,将1当量的ⅹ放入反应容器中,加入一定量二氯甲烷使化合物溶解,并使烧瓶处于-78℃环境。将1当量的六乙基亚磷酸胺溶解在一定量的二氯甲烷溶剂中,缓慢滴加进反应容器中之后,再常温搅拌反应隔夜。将得到的溶液先减压蒸馏除去溶剂之后,通过层析柱分离纯化之后,减压蒸馏旋干洗脱剂即可得到产物;其化学反应方程式如下所示:
其中,ar为苯环个数≥0的稠环、苯环并氮杂环、稠环并氮杂环、苯环接取代基并氮杂环或稠环接取代基并氮杂环,r为碳原子数≥12的直链或支链烷基。
优选地,所述1,2-稠环或稠环并氮杂环或稠环(含卤代基)并氮杂环二胺为1,2苯二胺、1,2萘二胺、2,3-二氨基酚嗪或4,5二氟-1,2苯二胺。
优选地,所述有机溶剂为冰乙酸,或冰乙酸与三氯甲烷,或冰乙酸与二氯甲烷的混合溶液。
优选地,所述洗脱剂为二氯甲烷,或甲苯,或二氯甲烷与乙酸乙酯的混合溶液。
本发明的新型氮杂稠环化合物拥有较大的共轭结构,有利于电子的传输,并且最大吸收波长在可见光领域,在有机场效应晶体管(ofet)、有机太阳能电池(opv)、有机发光二极管(oled)领域具有潜在的应用前景;因此,本发明还请求保护上述任一项所述的氮杂稠环化合物在制备光电材料中的应用。
本发明通过一种非常简单的方法合成出一种新型的、稳定的、溶解性好的氮杂稠环化合物。由于在共轭体系中引入了较长的烷基链,使得到的产物拥有在有机溶剂中拥有较好的溶解性,能够将其应用到复杂的器件工艺上。相比于其他的氮杂稠环化合物,本发明的合成方法极其的高效简便,且产物拥有较好的性能。
与现有技术相比,本发明具有以下有益效果:
(1)本发明通过一种简单的方法合成出一种新型的氮杂稠环化合物,所述化合物拥有共轭长度较长的大分子平面结构,使得化合物具有良好的稳定性和溶解性,同时通过在中心原子中引入了长烷基链增加了化合物的溶解性。
(2)相比于其他的氮杂稠环结构,本发明的合成方法具有较高的产率,能够获得的最终产物产率高于30%。
(3)本发明的新型氮杂稠环化合物拥有较大的共轭结构,有利于电子的传输,并且最大吸收波长在可见光领域,在有机场效应晶体管(ofet)、有机太阳能电池(opv)领域具有潜在的应用前景。
附图说明
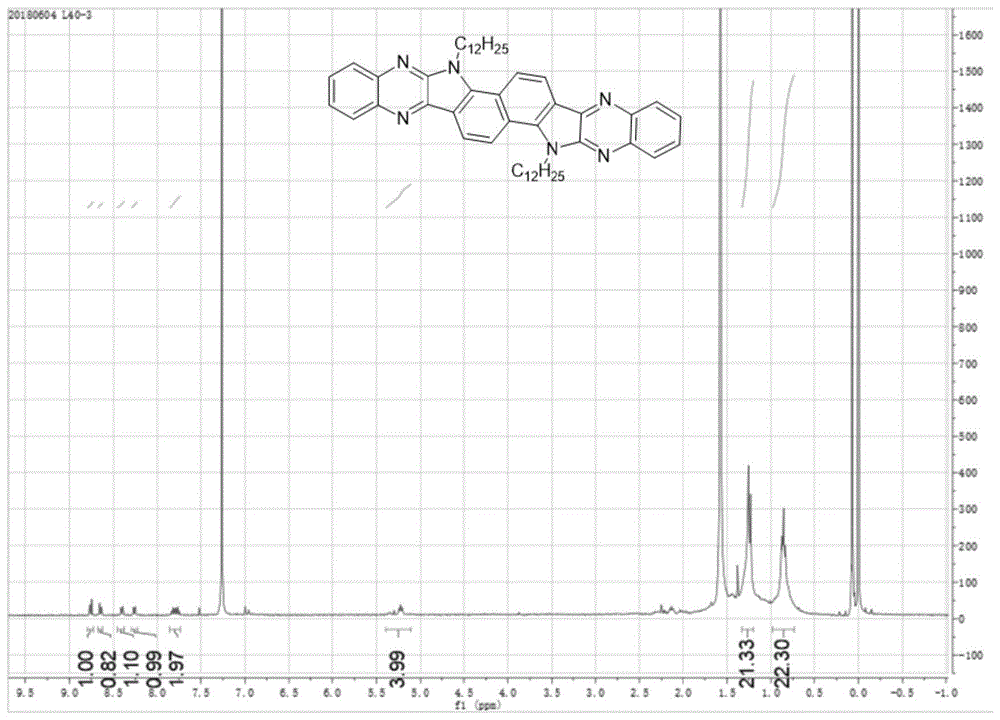
图1为化合物iii的1hnmr谱图,通过核磁共振氢谱可以证明合成的化合物为结构iii。
图2为化合物iii的质谱图,通过该谱图可以证明合成的化合物为结构iii。
图3为化合物iii的热重分析图,通过该谱图可以证明化合物iii拥有良好的热稳定性。
图4为化合物vi的1hnmr谱图,通过核磁共振氢谱可以证明合成的化合物为结构vi。
图5为化合物vi的质谱图,通过该谱图可以证明合成的化合物为结构vi。
图6为化合物vi的热重分析图,通过该谱图可以证明化合物vi拥有良好的热稳定性。
图7为化合物b的1hnmr谱图,通过核磁共振氢谱可以证明合成的化合物为结构b。
图8为化合物d的1hnmr谱图,通过核磁共振氢谱可以证明合成的化合物为结构d。
图9为化合物d的质谱图;通过该谱图可以证明合成的化合物为结构d。
图10为化合物f的1hnmr谱图,通过核磁共振氢谱可以证明合成的化合物为结构f。
图11为化合物f的质谱图;通过该谱图可以证明合成的化合物为结构f。
图12为化合物iii的紫外吸收曲线,通过曲线可以得知合成的化合物iii最大吸收波长为408nm,位于可见光范围。
图13为化合物vi的紫外吸收曲线,通过曲线可以得知合成的化合物vi最大吸收波长为430nm,位于可见光范围。
图14为化合物iii的循环伏安曲线。
图15为化合物vi的循环伏安曲线。
具体实施方式
以下结合说明书附图和具体实施例来进一步说明本发明,但实施例并不对本发明做任何形式的限定。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
除非特别说明,以下实施例所用试剂和材料均为市购。
本实验采用了瑞士bruker公司生产的avanceiii400m型液体核磁共振谱仪来检测产物的1hnmr、13cnmr谱图,溶剂为氘代氯仿(cdcl3),四甲基硅烷(tms)为内标。本实验采用瑞士bruker生产的solarix质量分析飞行质谱仪(maldi-tof)来表征分子量,利用shimadzu公司的uv-3600型紫外可见分光光度计来测定产物的紫外吸收光谱,采用1×1cm的石英比色皿作为样品池,采用上海辰华仪器有限公司的chi620e电化学分析仪。
实施例1
一种新型氮杂稠环化合物的制备方法,包括如下步骤:
在氮气环境下,将38mg1,2苯二胺(化合物i)与106mg3,8-双(2-十二烷基)-3,8-二氢吲哚[7,6g]吲哚-1,2,6,7-四酮(化合物ii)加入到单口烧瓶中(当量比为2比1),然后加入8ml冰乙酸,在120℃反应12h。将得到的溶液先减压蒸馏除去乙酸,再通过层析柱纯化,洗脱剂为二氯甲烷,分离提纯之后减压蒸馏旋干二氯甲烷即可得到黄色固体产物(化合物iii),得到化合物质量为48.7mg,产率为37%。所述产物的1hnmr谱图如图1所示,质谱图如图2所示,热重分析图如图3所示,通过该谱图可以证明化合物iii拥有良好的热稳定性。
上述反应的化学反应方程式如下所示:
其中化合物ii通过文献方法(j.mater.chem.a4,6940–6945(2016))制得。
实施例2
一种新型氮杂稠环化合物的制备方法,包括如下步骤:
在氮气环境下,将20mg1,2萘二胺(化合物iv)与52mg3,8-双(2-辛基十二烷基)-3,8-二氢吲哚[7,6g]吲哚-1,2,6,7-四酮(化合物v)加入到单口烧瓶中(当量比为2比1),然后加入4ml冰乙酸,在120℃反应4h。将得到的溶液先减压蒸馏除去乙酸,再通过层析柱纯化,洗脱剂为甲苯,分离提纯之后减压蒸馏旋干甲苯即可得到红色固体产物(化合物vi),得到化合物质量为30mg,产率为43.8%。所述产物的1hnmr谱图如图4所示,质谱图如图5所示,热重分析图如图6所示,通过该谱图可以证明化合物vi拥有良好的热稳定性。
上述反应的化学反应方程式如下所示:
其中化合物v通过文献方法(j.mater.chem.a4,6940–6945(2016))制得。
实施例3
一种新型氮杂稠环化合物的制备方法,包括如下步骤:
在氮气环境下,将8.7mg4,5二氟-1,2苯二胺(化合物a)与20mg3,8-双(2-辛基十二烷基)-3,8-二氢吲哚[7,6g]吲哚-1,2,6,7-四酮(化合物v)加入到单口烧瓶中,然后加入2ml冰乙酸和2ml氯仿,在75℃反应1.5h。将得到的溶液先减压蒸馏除去溶剂,再通过层析柱纯化,洗脱剂为二氯甲烷,分离提纯之后减压蒸馏旋干二氯甲烷即可得到黄色固体产物(化合物b),得到化合物质量为11.2mg,产率为45%。所述产物的1hnmr谱图如图7所示。
上述反应的化学反应方程式如下所示:
其中化合物v通过文献方法(j.mater.chem.a4,6940–6945(2016))制得。
实施例4
一种新型氮杂稠环化合物的制备方法,包括如下步骤:
在氮气环境下,将14mg2,3-二氨基酚嗪(化合物c)和55mg3,8-双(2-辛基十二烷基)-3,8-二氢吲哚[7,6g]吲哚-1,2,6,7-四酮(化合物v)加入到单口烧瓶中,然后加入2ml的冰乙酸和2ml氯仿混合溶剂,75℃反应4h。将得到的溶液先减压蒸馏除去溶剂,迅速的用二氯甲烷加乙酸乙酯分离提纯,蒸馏烘干后得到绿色固体质量为20mg,产率为30%。所述产物的1hnmr谱图如图8所示,质谱图如图9所示。
上述反应的化学反应方程式如下所示:
其中化合物v通过文献方法(j.mater.chem.a4,6940–6945(2016))制得。
实施例5
一种新型氮杂稠环化合物的制备方法,包括如下步骤:
在氮气环境下,将20mg化合物x放入圆底烧瓶中,加入5ml二氯甲烷使化合物溶解并处于-78℃环境。将4.93mg六乙基亚磷酸胺溶解在1ml的二氯甲烷溶剂中,缓慢滴加进反应容器中之后,再常温搅拌反应隔夜。将得到的溶液先减压蒸馏除去溶剂,通过层析柱分离纯化之后,再减压蒸馏旋干洗脱剂即可得到产物。产物为棕黑色固体,质量为18mg,产率为90%。所述产物的1hnmr谱图如图10所示,质谱图如图11所示。
上述反应的化学反应方程式如下所示:
性能测试
将化合物iii和化合物vi进行紫外吸收和循环伏安曲线测试,其检测结果如图12~图15所示:
化合物iii的紫外吸收曲线图如12所示,通过曲线可以得知合成的化合物ⅲ最大吸收波长为408nm,位于可见光范围。吸收峰尾波长为533nm,计算得出光学能级带隙为2.32ev。
化合物vi的紫外吸收曲线如图13所示,通过曲线可以得知合成的化合物ⅵ最大吸收波长为430,位于可见光范围。吸收峰尾波长为603nm,计算得出光学能级带隙为2.05ev。
化合物iii的循环伏安曲线如图14所示,通过曲线计算得到化合物ⅲ的lumo能级为-2.8665ev,homo能级为-5.1865ev。
化合物vi的循环伏安曲线如图15所示,通过曲线计算得到化合物ⅵ的lumo能级为-3.0625ev,homo能级为-5.1125ev。
综上所述,本发明通过一种简单的方法合成出一种新颖,并且具有较好的溶解性能的氮杂稠环化合物。相比于其他并苯结构,合成的并苯类似物具有较高的产率。用该简单方法合成的系列产物能够获得有效共轭长度达到十六个环的稳定结构,极其有利于电子的传输,并且最大吸收波长在可见光领域,在有机场效应晶体管(ofet)、有机太阳能电池(opv)、有机发光二极管(oled)领域具有很大的潜在应用前景。
- 还没有人留言评论。精彩留言会获得点赞!