杨小舟蛾谷胱甘肽S-转移酶MtGSTo1基因启动子与应用的制作方法
本发明属于基因工程技术领域,更具体地说,涉及一种杨小舟蛾谷胱甘肽s-转移酶mtgsto1基因启动子与应用。
背景技术:
在长期的演化中,植物和昆虫之间的密切的相互关系在很大程度上体现在植物次生物质的多样性上,昆虫演化了精确的适应性和抗性与之相适应。昆虫对植物次生物质的抗性主要包括代谢抗性、靶标抗性和行为抗性,一系列解毒酶系的出现是昆虫适应性的主要方式之一。谷胱甘肽s-转移酶(glutathiones-transferase,gsts)、羧酸酯酶(caboxyle-sterase,care)和细胞色素p450(cytochromep450,cyp450)是昆虫体内最重要的三大解毒酶系。其中gsts属于ii相代谢反应的重要转移酶,通过内源谷胱甘肽与有害的亲电子基团结合并排出体外或以非共价结合的蛋白质和疏水配基相结合,解除内源性或外源性毒物质的影响,从而起到解毒作用。
启动子是基因表达调控的重要顺式元件,在基因的表达调控中转录环节是最主要的调控位点,而启动子的调控在转录环节中又占据着十分重要的地位;植物次生物质可诱导昆虫体内gsts等解毒酶活性,有利于昆虫解毒。因此,有理由相信植物次生物质对gsts基因启动子很可能具有重要的调控功能。
技术实现要素:
发明目的:针对现有技术存在的上述问题,本发明的目的在于提供一种杨小舟蛾gsts基因启动子,在植物次生物质诱导下可在宿主细胞中被启动。本发明的另一目的在于提供杨小舟蛾gsts基因启动子序列的载体,可启动下游基因在宿主细胞中表达。本发明还有一目的是提供杨小舟蛾gsts基因启动子序列的载体的应用方法,可用于提高昆虫细胞对有毒物质抗性。
技术方案:为了解决上述问题,本发明所采用的技术方案如下:
一种杨小舟蛾谷胱甘肽s-转移酶mtgsto1基因启动子,其核苷酸序列如seqidno.1所示。
一种表达载体,含有所述的杨小舟蛾谷胱甘肽s-转移酶mtgsto1基因启动子。
一种宿主细胞,含有所述的表达载体。
优选的,所述的宿主细胞为昆虫细胞。
所述的杨小舟蛾谷胱甘肽s-转移酶mtgsto1基因启动子在宿主细胞中启动下游基因表达的应用。
所述的杨小舟蛾谷胱甘肽s-转移酶mtgsto1基因启动子在提高昆虫细胞对有毒物质抗性中的应用。
所述的杨小舟蛾谷胱甘肽s-转移酶mtgsto1基因启动子在启动表达mtgsto1基因中的应用
有益效果:相比于现有技术,本发明成功克隆出杨小舟蛾mtgsto1基因启动子,试验证实,该启动子可用于在宿主细胞中启动下游基因表达,尤其是在植物次生物质诱导下能够启动下游基因表达,可以为植物次生物质对于杨小舟蛾mtgsto1基因表达调控具体作用机制提供理论基础,为杨树食叶害虫新的防治技术提供理论依据和基因序列,也可以为培育出对有毒物质具有更高抗性的昆虫细胞或转基因家蚕提供备选基因。
附图说明
图1是杨小舟蛾幼虫dna电泳图;
图2是杨小舟蛾mtgsto1启动子pcr产物的电泳检测图;图中,m:marker,1:mtgsto1启动子;
图3pgl4.10-mtgsto1启动子是重组质粒的酶切验证结果图;图中,m:marker,1:pgl4.10,2:pgl4.10双酶切,6:pgl4.10-mtgsto1启动子双酶切;
图4是pgl4.10质粒图;
图5是杨小舟蛾mtgsto1基因启动子活性检测结果图;
图6是单宁胁迫下杨小舟蛾mtgsto1基因启动子活性检测结果图;
图7是槲皮素胁迫下杨小舟蛾mtgsto1基因启动子活性检测结果图;
图8是十三烷酮胁迫下杨小舟蛾mtgsto1基因启动子活性检测结果图。
具体实施方式
下面结合具体实施例对本发明进一步进行描述。以下实施例中未作具体说明的分子生物学实验方法,均可参照《分子克隆实验指南》(第三版)j.萨姆布鲁克一书中所列的方法或本领域的常规方法进行,或者按照试剂盒和产品说明书进行。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
供试昆虫:杨小舟蛾(micromelalophatroglodyta)采于江苏省南京市浦口区乌江镇,取回室内后在26土1℃恒温,h光照∶h黑暗=16∶8,相对湿度75%的条件下饲养。取幼虫在冰上解剖,于液氮中迅速冷冻后在-80℃冰箱中保存备用。
试剂的配置:
(1)lb液体培养基
用电子天平称取胰蛋白胨trypone10g,酵母提取物yeastextract5g,nacl10g,用naoh调节ph值至7.0,加ddh2o定容至1l,120℃高压灭菌20min。
(2)lb固体培养基
取1llb液体培养基,加入15g琼脂粉,120℃高压灭菌20min。
(3)50×tae电泳缓冲液100ml
tris24.2g,冰醋酸5.71ml,0.5medta(ph=8.0)10ml,加ddh2o定容至100ml。
(4)氨苄青霉素贮存液(100mg/ml)
称取100mg氨苄青霉素,溶于1mlddh2o中,-20℃保存。
(5)x-gal溶液配制
称取20mgx-gal,溶于1ml二甲基酰胺中,-20℃保存。
实施例1:杨小舟蛾谷胱甘肽s-转移酶基因启动子的克隆
1、引物的设计
根据染色体步移方法设计接头引物及通用引物,再根据成功克隆的gsts基因序列,利用primerpremier5软件设计两条特异引物,如下:
adaptorprimer1:
5′-gtaatacgactcactatagggcacgcgtggtcgacggcccgggctggt-3′,
nestedadaptorprimer2:5′-accagccc-3′,
common1:5′-gtaatacgactcactatagggc-3′,
common2:5′-actatagggcacgcgtggt-3′,
mtgsto11:5′-actaaaactgttctctcggcgtatggg-3′,
mtgsto12:5′-gggcagaatctcatagcgaatacacg-3′。
引物由上海捷瑞生物技术有限公司合成。
2、采用dnaisoreagent试剂盒(takara)提取杨小舟蛾基因组dna
(1)取解剖好的杨小舟蛾材料放入用液氮预冷的研钵中,加入适量dnaisoreagent液,用研磨杵快速研磨匀浆。(2)裂解液转移至离心管中,室温静置5min,4℃12000×g离心10min。(3)吸取上清液于新离心管中,再加入0.5ml无水乙醇,混匀1~3min,用枪头取沉淀至新的离心管中或4000×g离心2min倒出上清液。(4)沿管壁加入1ml75%乙醇,轻轻颠倒洗涤管壁,4℃12000×g离心5min弃乙醇。(5)室温干燥10min,加入灭菌水50μl,水溶10min,保存于-80℃备用。
在depc处理过的250ml的三角瓶中加入1×tae缓冲液20ml,再用电子天平称取0.2g琼脂糖,将其加入三角瓶中,混匀,微波炉加热至完全溶解。待溶液稍冷却至大约50℃时,加入2μl染料(gelstain)摇匀,倒入制胶板中,插上梳子。待胶液凝固,拔出梳子,将胶放入电泳槽,取1μlrna进行电泳检测。设定恒定电压140v,跑30min后,在自动凝胶成像系统上照相。取1μldna用于紫外分光光度仪检测dna的浓度和纯度。
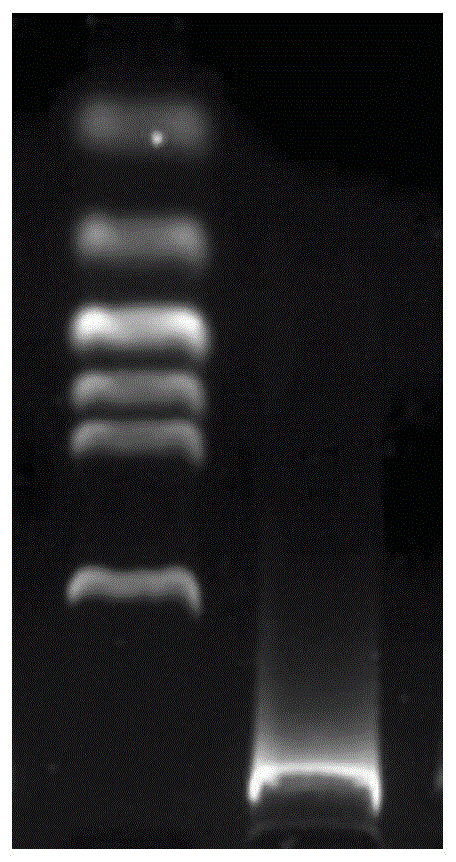
提取的杨小舟蛾dna的电泳结果如图1所示,有明显条带,且经nanodrop1000检测:rna的浓度为6722.3ng/μl,od260/od280值为2.04,od260/od230值为2.17。说明提取的杨小舟蛾的dna的质量较好,可用于后续的实验。
3、酶切
利用primerpremier5软件挑选合适的酶进行酶切反应。按下列组分配制反应液(25μl):2μl10×nebuffer,*xμldna,0.5μl酶,22.5-xddh2o。*x:取0.5μgtotaldna,根据dna浓度计算体积。反应程序:37℃forever。
4、纯化
酶切产物中加入等体积的tris-饱和酚,低速振荡5~10s。室温离心分层,吸取上清液于另一新离心管中。加入等体积的氯仿,低速振荡5~10s。室温离心分层,吸取上清液于另一新离心管中。加入两倍体积的95%乙醇,1/10体积的3mnaoac(ph=4.5),低速振荡5~10s。4℃14000×g离心15min,弃上清,用80%乙醇清洗沉淀。沉淀风干10min,加入适量灭菌水,水溶10min,保存于-80℃备用。
5、接头引物连接
(1)接头引物制备
adaptorprimer1和nestedadaptorprimer2长短引物各取10μl混合,放进刚煮开的水中,降到室温后再等两小时,即为接头引物(adaptorprimer)母液。
(2)接头引物连接
按下列组分配制反应液(8μl):1.6μl10×ligationbuffer,4μldna,1.9μladaptorprimer,0.5μlt4dnaligase。
反应程序:16℃ovemight;70℃5min。
(3)加入72μlddh2o,保存于-80℃备用。
6、pcr扩增
第一轮引物扩增:以连接了接头引物的dna为模版,进行pcr扩增。按下列组分配制pcr反应体系(25μl):1μltemplate,1μlprimer1,1μlcommon1,12.5μlmixex-tag,9.5μlddh2o。pcr反应程序:94℃5min;{94℃25s,72℃3min}7cycles;{94℃25s,67℃3min}32cycles;67℃7min;4℃forever。
第二轮引物扩增:第一轮产物稀释50倍作为第二轮产物模版;按下列组分配制pcr反应体系(25μl):1μltemplate2,1μlprimer2,1μlcommon2,12.5μlmixex-tag,9.5μlddh2o。pcr反应程序:94℃5min;{94℃25s,72℃3min}5cycles;{94℃25s,67℃3min}30cycles;67℃7min;4℃forever。
将反应液于琼脂糖凝胶电泳检测。
7、pcr产物的切胶回收、连接和转化
使用takara公司minibestagarosegeldnaextactionkitver3.0回收试剂盒(离心柱型)回收产物,步骤如下:
(1)从琼脂糖凝胶中切下含有目的片段的凝胶(尽量切除多余部分),精确称量重量,放入灭菌的1.5ml离心管中。(2)每100mg琼脂糖凝胶加入300μlbuffergm,37℃水浴放置3—5min,期间不断轻轻上下颠倒离心管,直至胶块完全融化。(3)观察溶解液颜色(如果不是黄色则加入3m醋酸钠(ph5.2)10μl混合至溶液恢复黄色)(4)将spincolumn安置于collectiontube上。(5)将溶液转移至spincolumn中12000rpm离心1min,弃滤液。(6)将700μl的bufferwb加入spincolumn,室温12000rpm离心30sec,弃滤液。(7)重复上步步骤,再次洗涤。(8)将spincolumn安置于collectiontube上,室温将spincolumn安置于新的1.5ml离心管上,在spincolumn膜的中央加入30μlddh2o,室温静置1min。(9)室温12000rpm离心1min,取1μl回收液用于紫外分光光度仪检测dna的浓度和纯度。
连接:将扩增产物与pdm19t载体连接。连接成分组成(5μl):2.5μlsolutionibuffer,*xμldna(回收),0.5μlpdm19tvector,(2-x)μlddh2o;*x:dna摩尔数为载体的三倍,根据dna浓度计算体积,回收的dna不足2μl用水补足。反应程序:16℃2h。
转化:(1)向5μl连接产物中加入100μl大肠杆菌感受态细胞,轻轻混匀,冰浴30min。(2)42℃水浴中热激60sec,然后立即置冰上3~5min。(3)加入800μlsoc培养基,37℃,150rpm摇床培养1h。(4)4000×g离心3min,弃上清,留大约150~200μl菌液,轻轻吹打沉淀,使其混匀。(5)蒋菌液涂在加有青霉素和x-gal的lb固体平板培养基上,37℃倒置过夜培养。(6)当培养皿上出现菌落后,挑取白色单菌落,用无菌牙签将单菌落挑入800μl含有amp的lb液体培养基中,37℃250转摇床培养。
将转化的菌液进行pcr检测,反应体系如下(25μl):1μl菌液,1μlprimer2,1μlcommon2,12.5μlmixex-tag,9.5μlddh2o;pcr反应程序:94℃5min;{94℃25s,72℃3min}5cycles;{94℃25s,67℃3min}30cycles;67℃7min;4℃forever。将反应液电泳检测,将具有符合目的片段大小的菌液送到生物公司测序。
将pcr反应产物在琼脂糖凝胶中进行电泳,结果如图2所示,成功克隆出1条杨小舟蛾gsts基因的启动子,命名为杨小舟蛾mtgsto1基因启动子,其序列长度为844(含atg)。利用生物信息学在线预测软件(http://www.gene-regulation.com/pub/programs/alibaba2/index.html.以及http://www.fruitfly.org/seq—tools/promoter.html),预测可能的转录起始位点,根据预测结果,mtgsto1基因启动子序列长度为596,如序列表中seqidno.1所示。
实施例2:杨小舟蛾谷胱甘肽s-转移酶基因启动子的活性验证
1、构建表达载体
1)根据成功克隆的启动子序列设计引物,经筛选,在正反向引物5′端加入了nhei和xhoi酶切位点,并加入保护碱基“aa”。用于杨小舟蛾gst基因启动子真核表达供体质粒构建的引物如下:
mtgsto1f:5′-aactcgagattactatagggcacgc-3′:
mtgsto1r:5′-aagctagcggtttgtaaatgtttttc-3′。
引物由上海捷瑞生物技术有限公司合成。
2)杨小舟蛾gsts基因启动子质粒提取:(1)从成功克隆的启动子平板培养基上挑选单菌落接种至1~4ml的含有抗生素的lb液体培养基中,37℃过夜培养(12~16h)。(2)取1~4ml的过夜培养菌液,12,000rpm离心2min,弃上清。(3)用250μl的solutioni(含rnasea)充分悬浮细菌沉淀。(4)加入250μl的solutionii轻轻上下翻转混合5~6次,使菌体充分裂解,形成透明溶液。(5)加入350μl的4℃预冷的solutioniii,轻轻上下翻转混合5~6次,直至形成紧实凝集块,然后室温静置2min。(6)室温12,000rpm离心10min,取上清。(7)将试剂盒中的spincolumn安置于collectiontube上。(8)将操作步骤6的上清液转移至spincolumn中,12,000rpm离心1min,弃滤液。(9)将500μl的bufferwa加入spincolumn中,12,000rpm离心30sec,弃滤液。(10)将700μl的bufferwb加入spincolumn中,12,000rpm离心30sec,弃滤液。(11)重复操作步骤(10)。(12)重新将spincolumn安置于collectiontube上,12,000rpm离心1min,除尽残留洗液。(13)将spincolumn安置于新的1.5ml的离心管上,在spincolumn膜的中央处加入50μl的elutionbuffer,室温静置1min。(14)12,000rpm离心1min洗脱dna,取1μl质粒溶液用于紫外分光光度仪检测dna的浓度和纯度。
3)pcr扩增:以质粒为模版,按下列组分配制pcr反应体系(25μl):*xμl质粒,1μl正向引物,1μl反向引物,12.5μlmixex-taq酶,(10.5-x)μlddh2o;*x:取1μg质粒dna,根据dna浓度计算体积。pcr反应程序:94℃5min;{94℃30s,62℃30s,72℃1min30s}25cycles;72℃10min;4℃forever。将反应液于琼脂糖凝胶电泳检测。
4)pcr产物切胶回收、连接、转化
dna目的片段回收方法同实施例1。
加酶切位点启动子质粒提取方法同上。
双酶切:提取的质粒与pgl4.10载体利用nhei和xhoi内切酶进行双酶切,按下列组分配制酶切反应体系(20μl):*xμl质粒,1μlxhoi,1μlnhei,(18-x)μlddh2o;*x:取1μg质粒dna,根据dna浓度计算体积。酶切反应程序:37℃2h;
连接:用同样的酶双酶切处理目标载体pgl4.10用于连接,连接成分组成(10μl):1μl10×ligationbuffer,*xμldna(酶切产物),0.5μlpgl4.10vector,1μlt4dnaligase,(7.5-x)μlddh2o;*x:dna摩尔数为载体的三倍,根据dna浓度计算体积。反应程序:16℃2h。
转化方法同实施例1。
重组质粒的验证:经测序验证后,提取质粒,进行酶切反应。
经测序和酶切验证(图3、图4),pgl4.10与mtgsto1基因启动子成功构建重组质粒,可用于后续细胞转染实验。
2、重组质粒对昆虫sf9细胞的转染
1)昆虫sf9细胞的贴壁培养:用含10%fbs(胎牛血清)和抗生素的sf-900iisfm细胞培养基将昆虫sf9细胞培养于细胞培养瓶,27℃恒温生化培养箱中避光培养,根据细胞状态2~3d传代培养。
2)昆虫sf9细胞的冻存:生长状态良好且传代次数少的未变形细胞需冻存留种,轻轻吹下贴壁长满的细胞,5000g离心10min,细胞沉淀用预冷细胞冻存液(含10%dmso的全细胞培养基)悬浮。细胞总量可在离心前使用计数板计数,也可根据培养瓶底面积估算,使细胞终浓度约为1×107cell/ml。将细胞冻存液分装到冰浴的细胞冻存管中,4℃,30min;-20℃,30min;-80℃,30min;最后将冻存管置于液氮中保存备用。
3)昆虫sf9细胞的复苏:将培养基加入培养瓶,从液氮中取出细胞冻存管后迅速27℃水浴,晃动至还有少量冰残留时取出,用酒精消毒外壁,将冻存液吸取至培养瓶中。27℃恒温生化培养箱中避光培养,细胞贴壁后换液(一般低于30min),培养24h后再换液,测细胞活力,成功复苏的细胞活力大于75%。
4)使用pureyieldtmplasmidmidiprepsystem(promega)提取去内毒素质粒:(1)从重组质粒平板培养基上挑选单菌落接种至50~100ml的含有抗生素的lb液体培养基中,37℃过夜培养(12~16h)。(2)取过夜培养的菌液5000g离心10min,弃上清。(3)加入3mlcellresuspensionsolution充分悬浮细菌沉淀。(4)加入3mlcelllysissolution轻轻上下翻转混合3~5次,室温静置3min,使菌体充分裂解,形成透明溶液。(5)加入5ml4℃预冷的neutralizationsolution轻轻上下翻转混合5~10次。(6)室温15000g离心15min。(7)将蓝色clearingcolumn置于白色bindingcolumn上,安置于真空抽提装置。(8)取上清液于clearingcolumn,利用真空装置使其通过,弃clearingcolumn。(9)加5mlendotoxinremovalwash于bindingcolumn,利用真空装置使其通过。(10)加20mlcolumnwashsolution于bindingcolumn,利用真空装置使其通过。(11)利用真空装置干燥膜30~60sec。(12)擦去bindingcolumn上的酒精,加入400~600μlnuclease-freewater静置1min,用新的ep管收集。
5)采用脂质体转染法invitrogencellfectionriireagent转染细胞
(1)铺板:用1ml含双抗和血清的培养基清洗将要传代的细胞,加入适量的含双抗和血清培养基将细胞吹起后分装至96孔板静置15min以上(每孔约9×104个细胞)。(2)配制转染混合液(单孔用量):a液:1μl脂质体,10μl无血清无抗生素培养基;室温静置30min。b液:200ng目标质粒,20ngprl-tk,10μl无血清无抗生素培养基;将a液加到b液中混匀,室温静置30min。(3)转染:将完成铺板的细胞中培养基吸出,加入80μl无血清无抗生素的培养基,再加入20μl转染混合液,置于27℃培养5h后换上含血清含双抗的培养基150μl,置于27℃培养48h后进行双荧光检测。
将启动子重组质粒利用脂质体转染法转到昆虫sf9细胞中表达,转染48h后利用双荧光检测荧光素酶的活性,萤火虫荧光检测值(luc)与海肾荧光检测值(rluc)比值即为最终结果,并利用方差分析各启动子活性相对于载体对照的显著性差异(***表示p<0.001,*表示p<0.05),如图5所示。结果表明,mtgsto1启动子具有非常强的活性,与载体相比有极强的显著性。
实施例3植物次生物质对mtgsto1基因启动子重组质粒的诱导表达
主要方法同实施例2,在细胞转染过程中,27℃培养5h后换上含血清含双抗的培养基时加入10μl诱导药物,本实施例共选取单宁、槲皮素和2-十三烷酮三种植物次生物质对杨小舟蛾mtgsto1基因启动子进行表达诱导,用ddh2o溶解单宁、丙酮溶解槲皮素和2-十三烷酮后,分别配制为0.01、0.1和1mg/ml三个浓度的药剂,其中单宁以ddh2o为对照,槲皮素和2-十三烷酮以丙酮为对照。
1)pgl4.10和杨小舟蛾mtgsto1基因启动子重组质粒转染sf9细胞过程中加入不同浓度的单宁进行表达诱导,并对萤光素酶检测结果进行方差分析(图6)。结果显示,在单宁胁迫下,杨小舟蛾mtgsto1基因启动子表达受到一定的诱导,mtgsto1基因启动子在0.01和0.1mg/ml浓度单宁胁迫下表达量显著上升(p<0.05)。
2)pgl4.10和杨小舟蛾mtgsto1基因启动子重组质粒转染sf9细胞过程中加入不同浓度的槲皮素进行表达诱导,并对萤光素酶检测结果进行方差分析(图7)。结果显示,mtgsto1基因启动子在各浓度槲皮素胁迫下,活性呈现一定的下降(p<0.05),表明槲皮素抑制了其启动子在sf9细胞中的表达。
3)pgl4.10和杨小舟蛾mtgsto1基因启动子重组质粒转染sf9细胞过程中加入不同浓度的2-十三烷酮进行表达诱导,并对萤光素酶检测结果进行方差分析(图8)。结果显示,mtgsto1基因启动子在一定浓度2-十三烷酮胁迫下表达受到了显著性的抑制作用。
以上说明对本发明而言只是说明性的,而非限制性的,本领域普通技术人员理解,在不脱离所附权利要求所限定的精神和范围的情况下,可做出许多修改、变化或等效,但都将落入本发明的保护范围。
序列表
<110>南京林业大学
<120>杨小舟蛾谷胱甘肽s-转移酶mtgsto1基因启动子与应用
<130>100
<160>9
<170>siposequencelisting1.0
<210>1
<211>596
<212>dna
<213>micromelalophatroglodyta
<400>1
gcttgcatgcctgcaggtcgacgattaactcgagattactatagggcacgcgtggtcgac60
ggcccgggctggtatctttcaattaatacagtcctttactgaaaaacgtcacttcatggt120
acagtgtggtgacactgtatcaaagctctgccctatcttcgccagagtcccgcagggaag180
cgtatctggccccccactgtacctgctgtacacttctgatttccccactaccgctgaagt240
aacaattggcacttttgctgacgatactgccataatagcacgacacagtgacccttcaat300
tatatttcatcgtcaacgtcacctcatttcattaaaacagtgcataatgtgtaattagtg360
atggatttttatgcattgtggacctgtgacagtggaaaattctgaaaacccgaacaataa420
gtgtagtgtgcggttcgccattagttttttgttggtgaagtggacccttgtgtccgactt480
tgtgaagttgccgccaaaactccgagaatcattttagcttgccatcacatgctataatgt540
ttcgaaaattacataagaagaaatattaaataagacgaggcggtatagtccacgac596
<210>2
<211>48
<212>dna
<213>adaptorprimer1引物序列(artificial)
<400>2
gtaatacgactcactatagggcacgcgtggtcgacggcccgggctggt48
<210>3
<211>8
<212>dna
<213>nestedadaptorprimer2引物序列(artificial)
<400>3
accagccc8
<210>4
<211>22
<212>dna
<213>common1引物序列(artificial)
<400>4
gtaatacgactcactatagggc22
<210>5
<211>19
<212>dna
<213>common2引物序列(artificial)
<400>5
actatagggcacgcgtggt19
<210>6
<211>27
<212>dna
<213>mtgsto11引物序列(artificial)
<400>6
actaaaactgttctctcggcgtatggg27
<210>7
<211>26
<212>dna
<213>mtgsto12引物序列(artificial)
<400>7
gggcagaatctcatagcgaatacacg26
<210>8
<211>25
<212>dna
<213>mtgsto1f引物序列(artificial)
<400>8
aactcgagattactatagggcacgc25
<210>9
<211>26
<212>dna
<213>mtgsto1r引物序列(artificial)
<400>9
aagctagcggtttgtaaatgtttttc26
- 还没有人留言评论。精彩留言会获得点赞!