一种阿维巴坦中间体、制备方法及其应用与流程
本发明属于药物合成领域,具体涉及一种阿维巴坦中间体[(1r,2s,5r)-2-(氨基羰基)-7-氧代-1,6-二氮杂双环[3.2.1]辛-6-基]硫酸铵、制备方法及其应用。
背景技术:
阿维巴坦(avibactam)是一种新型的非β-内酰胺结构的β-内酰胺酶抑制剂,与广谱头孢菌素头孢他啶(ceftazidime)联合用于治疗复杂性腹腔内感染(ciai)和复杂性尿路感染(cuti),目前该联合用药已获得fda批准上市,商品名avycaz。与其他抗菌素的联用(如头孢洛林酯、噻肟单酰胺菌素等)正处于临床研究中。阿维巴坦较之前上市的3个β-内酰胺酶抑制剂-克拉维酸、舒巴坦、三唑巴坦-作用更强,范围更广,对a类、c类和部分d类β-内酰胺酶抑制作用显著。
阿维巴坦具有二氮杂二环辛烷骨架,与经典的β-内酰胺酶抑制剂结构不同,它自身结构可经逆反应恢复,具有长效的抑制酶作用。另外,经典的β-内酰胺酶抑制剂对c类酶不具有或具有微弱的抑制作用,但阿维巴坦抑制c类酶作用显著,抑酶谱更广。阿维巴坦在临床上以其钠盐形式应用,其化学名为硫酸单[(1r,2s,5r)-2-氨基羰基-7-氧代-1,6-氮杂二环[3.2.1]辛-6-基酯钠盐,具体结构如下:
关于阿维巴坦的合成,有多篇文献报道,例如:cn103649051b、cn105758367a、cn106699756a、cn106699756a等。
目前的工艺中,大多是采用三氧化硫三甲胺络合物或so3.dmf等这些三氧化硫的配合物与(1r,2s,5r)-6-羟基-7-氧代-1,6-二氮杂环[3.2.1]辛烷-2-甲酰胺反应,然后再与四丁基铵盐成盐。然而三氧化硫三甲胺络合物或so3.dmf等这些三氧化硫的配合物价格均较为昂贵,给该化合物的工业化生产带来了巨大的阻力。
现有的有些工艺中,采用氯磺酸为磺化试剂,该磺化试剂价格相对较低。但是生成的磺酸化合物无法与四丁基铵盐直接成盐,需要首先利用碳酸氢铵等试剂转换为铵盐,得到的铵盐才可以与四丁基铵盐反应,这样就需要两步反应,操作繁琐,且收率较低。
技术实现要素:
本发明提供了一种阿维巴坦中间体[(1r,2s,5r)-2-(氨基羰基)-7-氧代-1,6-二氮杂双环[3.2.1]辛-6-基]硫酸铵(iii)以及其制备方法,该方法步骤少,操作简便。
本发明同时公开了一种利用上述中间体制备阿维巴坦的方法。
一种阿维巴坦中间体,包括下式所示结构中的一种或多种:
a为三烷基胺,吡啶,烷基吡啶,n-烷基哌啶,二烷基苯胺或二烷基苄胺,其中:所述三烷基胺中烷基为c5-8的烷基,烷基吡啶中烷基为c1-4的烷基,n-烷基哌啶中烷基为c1-4的烷基,二烷基苯胺中烷基为c1-4的烷基,二烷基苄胺中烷基为c1-4的烷基。即所述a+可以来源于三烷基胺,吡啶,烷基吡啶,n-烷基哌啶,二烷基苯胺或二烷基苄胺或者其他反应后能够形成三烷基胺,吡啶,烷基吡啶,n-烷基哌啶,二烷基苯胺或二烷基苄胺的化合物。
作为优选,所述a为苄基二甲胺、4-甲基吡啶、三戊胺。
一种阿维巴坦中间体的制备方法,包括:在碱剂存在下,化合物ii((1r,2s,5r)-6-羟基-7-氧代-1,6-二氮杂环[3.2.1]辛烷-2-甲酰胺)与磺化试剂反应,然后再与碱剂反应,得到所述阿维巴坦中间体([(1r,2s,5r)-2-(氨基羰基)-7-氧代-1,6-二氮杂双环[3.2.1]辛-6-基]硫酸铵);
所述化合物ii结构如下:
所述碱剂为三烷基胺,吡啶,烷基吡啶,n-烷基哌啶,二烷基苯胺或二烷基苄胺,其中:所述三烷基胺中烷基为c5-8的烷基,烷基吡啶中烷基为c1-4的烷基,n-烷基哌啶中烷基为c1-4的烷基,二烷基苯胺中烷基为c1-4的烷基,二烷基苄胺中烷基为c1-4的烷基。
作为优选,所述磺化试剂为氯磺酸、硫酸中的一种或多种。进一步优选为氯磺酸。
作为优选,所述碱剂为苄基二甲胺、4-甲基吡啶、三戊胺中的一种或多种。
作为优选,所述碱剂与所述化合物ii的摩尔比为(2~6):1。进一步优选为(3~4):1;更进一步优选为4:1。
作为优选,所述磺化试剂与所述化合物ii的摩尔比为(1~3):1。进一步优选为(1.5~2):1;更进一步优选为所述磺化试剂与所述化合物ii的摩尔比为2:1。
作为优选,反应温度为-10~30℃。进一步优选的反应温度为-5~10℃,更进一步优选的反应温度为0~5℃。
作为优选,反应溶剂为二氯甲烷、氯仿、四氢呋喃、乙腈、丙酮、乙酸乙酯等。
作为优选,所述碱剂为苄基二甲胺;
所述碱剂与所述化合物ii的摩尔比为(3~4):1;所述磺化试剂与所述化合物ii的摩尔比为(1.5~2):1;反应温度为-5~10℃。
磺化反应的后产物直接与碱剂进行反应,反应完成后,通过简单的萃取即可将产物[(1r,2s,5r)-2-(氨基羰基)-7-氧代-1,6-二氮杂双环[3.2.1]辛-6-基]硫酸铵从体系中萃取出来,在萃取前,可以选择预先将反应溶剂去除。萃取剂可以选择二氯甲烷等。萃取后,合并有机相,去除萃取剂,加入异丙醚,既可以析出高纯度的产品:[(1r,2s,5r)-2-(氨基羰基)-7-氧代-1,6-二氮杂双环[3.2.1]辛-6-基]硫酸铵。
一种上述任一技术方案所述阿维巴坦中间体制备阿维巴坦的方法,将所述阿维巴坦中间体用钠离子处理得到阿维巴坦。
作为优选,所述钠离子来源于异辛酸纳。
作为优选,所述化合物ii由(1r,2s,5r)-6-苄氧基-7-氧代-1,6-二氮杂环[3.2.1]辛烷-2-甲酰胺直接加氢还原得到。(1r,2s,5r)-6-苄氧基-7-氧代-1,6-二氮杂环[3.2.1]辛烷-2-甲酰胺可以利用钯碳作为催化剂进行加氢还原,反应后,直接过滤洗涤,过滤得到的滤液可以直接进行上述反应,即可以节省上述反应的反应溶剂用量,也节省了加氢还原后的后处理操作。
催化加氢采用的反应溶剂可以为二氯甲烷、氯仿、四氢呋喃、乙腈、丙酮、乙酸乙酯等。反应温度为20~40℃,进一步优选为所述的氢化温度为25~35℃。反应时间为3~5h。
作为优选,一种制备阿维巴坦的方法,包括:以(1r,2s,5r)-6-苄氧基-7-氧代-1,6-二氮杂环[3.2.1]辛烷-2-甲酰胺为原料。通过氢化、磺化制备阿维巴坦中间体[(1r,2s,5r)-2-(氨基羰基)-7-氧代-1,6-二氮杂双环[3.2.1]辛-6-基]硫酸铵,再用钠离子交换成钠盐。该方法反应条件温和,中间减少用铵盐成盐步骤,操作简便。
一种阿维巴坦中间体[(1r,2s,5r)-2-(氨基羰基)-7-氧代-1,6-二氮杂双环[3.2.1]辛-6-基]硫酸铵(iii)的制备办法,该方法包含了以下步骤:
(1)化合物i在钯炭的催化下脱苄生成化合物ii;
(2)化合物ii在碱剂的存在下与磺化试剂反应再和碱反应生成化合物iii;
(3)化合物iii用钠离子处理成化合物iv。
以碱剂为苄基二甲胺为例,其反应过程为:
与传统方法相比,本发明采用适当碱性的碱剂,代替传统的碱剂(例如代替碳酸氢铵),可以直接与磺化试剂一步进行反应,不需要另外的成盐化操作,得到的[(1r,2s,5r)-2-(氨基羰基)-7-氧代-1,6-二氮杂双环[3.2.1]辛-6-基]硫酸铵可以直接与钠盐反应,后处理简单,更适于工业化大量生产。
同时,本发明配合新的碱剂,我们可以采用价格更为经济的磺化试剂进行反应,从成本上大大降低了企业的生产压力。
本发明的碱剂在作为反应物的同时,也同时兼具缚酸剂的功能,不需要额外的添加缚酸剂,避免了缚酸剂杂质的残留,降低了后处理难度。
附图说明
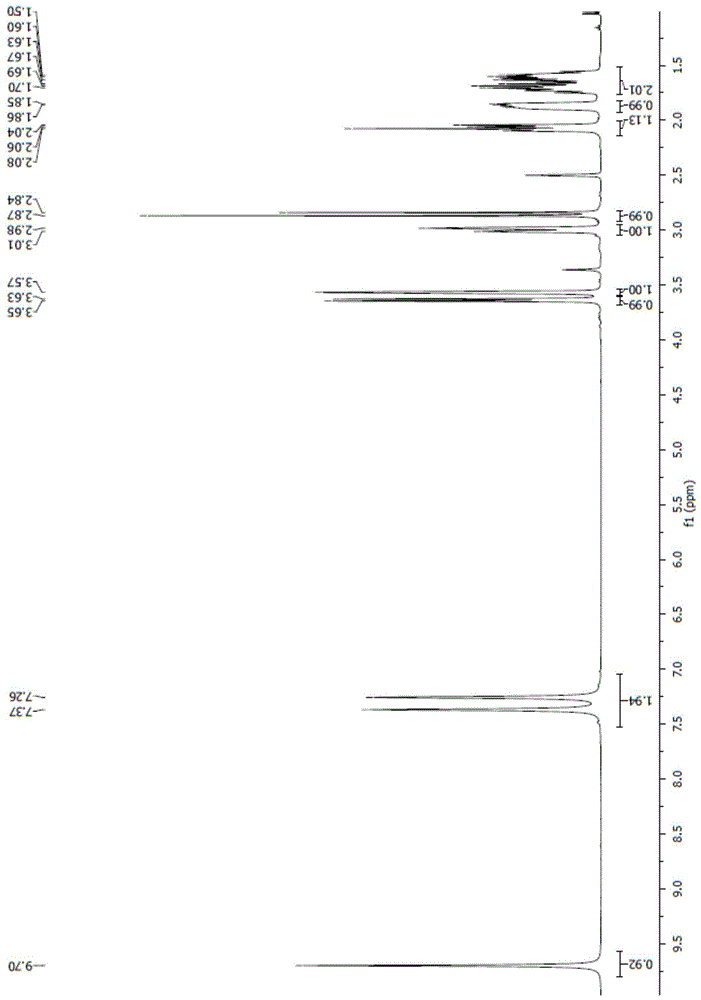
图1为实施例得到的化合物ii的核磁谱图-氢谱;
图2为实施例得到的化合物ii的核磁谱图-碳谱;
图3为实施例得到的化合物iii的核磁谱图-氢谱;
图4为实施例得到的化合物iii的核磁谱图-碳谱。
具体实施方式
实施例1:
反应瓶内投入化合物i((1r,2s,5r)-6-苄氧基-7-氧代-1,6-二氮杂环[3.2.1]辛烷-2-甲酰胺)5g(18mmol),丙酮50ml,钯炭0.5g,用氮气置换至少3次,用氢气置换3次,25~35℃下,通入氢气保温3h,tlc检测反应完全,用氮气置换3次,抽滤,用乙腈淋洗滤饼,滤液降温到0℃,加入苄基二甲胺9.7g(72mmol),滴加氯磺酸4.2g(36mmol)。保温2h,反应完全,反应液减压至干,加入二氯甲烷50ml,纯水30ml,分相,水相用20ml二氯甲烷提取一次,合并有机相,减压至干,加入异丙醚20ml,有固体析出,抽滤,用异丙醚淋洗,35℃烘料,得化合物iii([(1r,2s,5r)-2-(氨基羰基)-7-氧代-1,6-二氮杂双环[3.2.1]辛-6-基]硫酸苄基二甲铵)5.8g,收率80%,含量>99%。其中化合物ii核磁如下:1hnmr(400mhz,dmso)δ9.70(s,1h),7.31(d,j=44.8hz,2h),3.64(d,j=7.2hz,1h),3.57(s,1h),3.00(d,j=11.5hz,1h),2.86(d,j=11.6hz,1h),2.07(dd,j=13.8,7.1hz,1h),1.92–1.82(m,1h),1.76–1.52(m,2h),图谱见图1。13cnmr(101mhz,dmso)δ172.19(s),167.51(s),59.50(s),59.11(s),47.64(s),40.54(s),40.42(d,j=21.0hz),40.15(s),40.15–39.61(m),39.49(s),39.28(s),20.84(s),18.38(s),图谱见图2。化合物iii核磁如下:1hnmr(400mhz,dmso)δ7.57–7.15(m,7h),4.26(s,2h),3.98(s,1h),3.68(d,j=6.0hz,1h),2.96(dd,j=37.1,11.6hz,2h),2.72(s,6h),2.05(d,j=7.2hz,1h),1.83(d,j=3.5hz,1h),1.63(t,j=9.7hz,2h),图谱见图3。13cnmr(101mhz,dmso)δ172.00(s),166.58(s),131.35(s),131.01(s),130.01(s),129.37(s),60.18(d,j=21.6hz),59.91–59.79(m),58.06(s),47.40(s),42.34(s),21.00(s),18.54(s),谱图见图4。
反应瓶内投入化合物iii5.0g,20~35℃加入乙醇25ml,20~35℃溶清,滴加异辛酸纳乙醇溶液,有固体析出,20~35℃保温2h,抽滤,用无水乙醇淋洗,烘干。得到化合物iv3.3g,收率92%,含量:99%。
实施例2:
反应瓶内投入化合物i5g(18mmol),丙酮50ml,钯炭0.5g,用氮气置换至少3次,用氢气置换3次,25~35℃下,通入氢气保温3h,tlc检测反应完全,用氮气置换3次,抽滤,用丙酮淋洗滤饼,滤液降温到0℃,加入苄基二甲胺9.7g(72mmol),滴加硫酸3.5g(36mmol)。保温2h,反应完全。反应液减压至干,加入二氯甲烷50ml,纯水30ml,分相,水相用20ml二氯甲烷提取一次,合并有机相,减压至干,加入异丙醚20ml,有固体析出,抽滤,用异丙醚淋洗,35℃烘料,得化合物iii5.9g,收率83%,含量>99%。
反应瓶类投入化合物iii5.0g,20~35℃加入乙醇25ml,20~35℃溶清,滴加异辛酸纳乙醇溶液,有固体析出,20~35℃保温2h,抽滤,用无水乙醇淋洗,烘干。得到化合物iv3.4g,收率95%,含量:99%。
实施例3:
反应瓶内投入化合物i5g(18mmol),丙酮50ml,钯炭0.5g,用氮气置换至少3次,用氢气置换3次,25~35℃下,通入氢气保温5h,tlc检测反应完全,用氮气置换3次,抽滤,用丙酮淋洗滤饼,滤液降温到0℃,加入苄基二甲胺9.7g(72mmol),滴加硫酸3.5g(36mmol)。保温2h,反应完全。反应液减压至干,加入二氯甲烷50ml,纯水30ml,分相,水相用20ml二氯甲烷提取一次,合并有机相,减压至干,加入乙醇25ml,20~35℃溶清,滴加异辛酸纳乙醇溶液,有固体析出,20~35℃保温2h,抽滤,用无水乙醇淋洗,烘干。得到化合物iv4.0g,收率77%,含量:99%。
实施例4:
反应瓶内投入化合物i5g(18mmol),四氢呋喃50ml,钯炭0.5g,用氮气置换至少3次,用氢气置换3次,25~35℃下,通入氢气保温4h,tlc检测反应完全,用氮气置换3次,抽滤,用四氢呋喃淋洗滤饼,滤液降温到0℃,加入4-甲基吡啶6.7g(72mmol),滴加氯磺酸3.15g(27mmol)。保温2h,反应完全,反应液减压至干,加入二氯甲烷50ml,纯水30ml,分相,水相用20ml二氯甲烷提取一次,合并有机相,减压至干,20~35℃加入乙醇25ml,20~35℃溶清,滴加异辛酸纳乙醇溶液,有固体析出,20~35℃保温2h,抽滤,用无水乙醇淋洗,烘干。得到化合物iv4.2g,收率80.7%,含量:99%。
实施例5:
反应瓶内投入化合物i5g(18mmol),丙酮50ml,钯炭0.5g,用氮气置换至少3次,用氢气置换3次,25~35℃下,通入氢气保温3h,tlc检测反应完全,用氮气置换3次,抽滤,用丙酮淋洗滤饼,滤液降温到0℃,加入4-甲基吡啶6.7g(72mmol),滴加氯磺酸3.15g(27mmol)。保温2h,反应完全,反应液减压至干,加入二氯甲烷50ml,纯水30ml,分相,水相用20ml二氯甲烷提取一次,合并有机相,减压至干,20~35℃加入乙醇25ml,20~35℃溶清,滴加异辛酸纳乙醇溶液,有固体析出,20~35℃保温2h,抽滤,用无水乙醇淋洗,烘干。得到化合物iv4.1g,收率78.8%,含量:99%。
实施例6:
反应瓶内投入化合物i5g(18mmol),丙酮50ml,钯炭0.5g,用氮气置换至少3次,用氢气置换3次,25~35℃下,通入氢气保温3h,tlc检测反应完全,用氮气置换3次,抽滤,用丙酮淋洗滤饼,滤液降温到0℃,加入三戊胺16.4g(72mmol),滴加氯磺酸3.15g(27mmol)。保温2h,反应完全,反应液减压至干,加入二氯甲烷50ml,纯水30ml,分相,水相用20ml二氯甲烷提取一次,合并有机相,减压至干,20~35℃加入乙醇25ml,20~35℃溶清,滴加异辛酸纳乙醇溶液,有固体析出,20~35℃保温2h,抽滤,用无水乙醇淋洗,烘干。得到化合物iv4.0g,收率77%,含量:99%。
对比例1
按照实施例1的条件,将苄基二甲胺替换为等摩尔量的碳酸氢铵,按照相同的后处理方法,最后几乎没有得到任何[(1r,2s,5r)-2-(氨基羰基)-7-氧代-1,6-二氮杂双环[3.2.1]辛-6-基]硫酸铵,可能是几乎全部的氯磺酸被碳酸氢铵直接消耗成盐。
对比例2
将实施例3、实施例4苄基二甲胺9.7g(72mmol)替换为72mmol的二甲基甲酰胺,其他条件同实施例3、实施例4,在与硫酸或氯磺酸反应完成后,通过检测发现生成的化合物为(1r,2s,5r)-6-磺酸基-7-氧代-1,6-二氮杂环[3.2.1]辛烷-2-甲酰胺。且与异辛酸纳乙醇溶液反应后,最后没有得到最终的产品化合物iv。这也证明,利用氯磺酸、硫酸等做磺化试剂时,其得到的(1r,2s,5r)-6-磺酸基-7-氧代-1,6-二氮杂环[3.2.1]辛烷-2-甲酰胺无法直接与异辛酸纳反应。
对比例3
反应瓶内投入化合物i5g(18mmol),四氢呋喃50ml,钯炭0.5g,用氮气置换至少3次,用氢气置换3次,25~35℃下,通入氢气保温4h,tlc检测反应完全,用氮气置换3次,抽滤,用四氢呋喃淋洗滤饼,滤液降温到0℃,加入二甲基甲酰胺(72mmol),滴加氯磺酸3.15g(27mmol)。保温2h,反应完全,将体系液加入到碳酸氢铵(180mmol)水溶液(40ml)中,保持ph值大于6,搅拌15min,加入二氯甲烷,分层,水相用二氯甲烷洗涤,合并有机相,然后加入四丁基硫酸氢铵(27mmol)在二氯甲烷(100ml)中的溶液,搅拌15min。分离各层,用二氯甲烷(50ml)萃取水层。有机相减压蒸干,20~35℃加入乙醇25ml,20~35℃溶清,滴加异辛酸纳乙醇溶液,有固体析出,20~35℃保温2h,抽滤,用无水乙醇淋洗,烘干。得到化合物iv2.8g,收率55%。明显低于本发明的收率。
- 还没有人留言评论。精彩留言会获得点赞!
- Josiphos类手性二茂铁膦配体的合成方法
- (S)-1-(2-羟基-1-苯乙基)硫脲修饰的Mn-Anderson型杂多酸催化剂、制备方法及其应用
- (S)-1-(1-苯乙基)硫脲修饰的Cr-Anderson型杂多酸催化剂、制备方法及其应用
- (R)-1-(1-苯乙基)硫脲修饰的Cr-Anderson型杂多酸催化剂、制备方法及其应用
- 以机械增强来提高纤维素生物质的微生物转化的方法
- (S)-1-(2-羟基-1-苯乙基)硫脲修饰的Cr-Anderson型杂多酸催化剂、制备方法及其应用
- 一种微电解-Fenton内循环固定床反应器的制造方法
- 一种光学活性中间体n-叔丁氧羰基-2-氨基-8-壬烯酸二环己胺盐的合成方法
- 一种光学活性中间体n-叔丁氧羰基-2-氨基-8-壬烯酸二环己胺盐的合成方法
- 一种仿过氧化物酶材料及其应用