一种甲苯咪唑C晶型的制备新方法与流程
本发明属于化学或药物化学领域,具体涉及甲苯咪唑c晶型的制备方法。
背景技术:
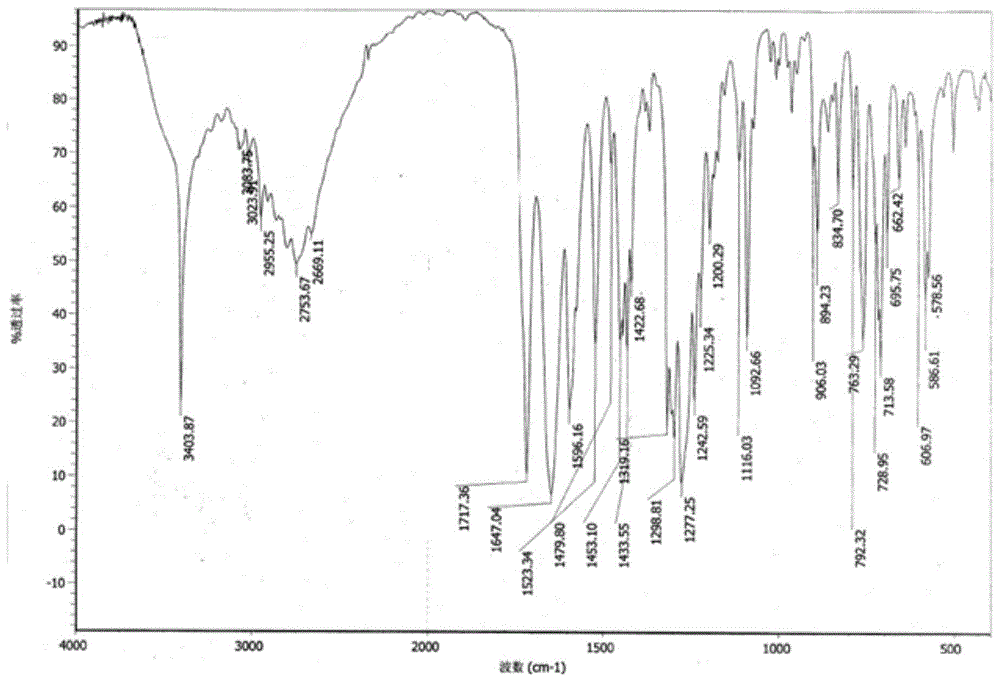
:甲苯咪唑(mebendazole)是比利时janssen公司首先研制生产的广谱驱虫药,它有3种不同晶型,据有关药理驱虫试验结果证明,c型为有效晶型,a型为无效晶型,b型目前数据不详。甲苯咪唑,化学名为:(5-苯甲酰基-1-h-苯并咪唑-2-基)-氨基甲酸甲酯。其理化性质为白色结晶粉末,无臭、无味;溶于冰乙酸、无机酸,微溶于甲醇、二氧六环、四氢呋喃、二甲基甲酰胺,几乎不溶于水;产品熔点大于280℃。临床表明,c型甲苯咪唑具有广谱驱肠道寄生虫药的特性,对线虫、绦虫有强大的驱虫作用,如蛲虫、蛔虫、十二指肠钩虫、美洲钩虫、类园线虫、绦虫等。剂量适宜时,90%以上的患者(即使有严重的或混合感染的)都可以除尽肠虫。2002年,刘祥宜等(江苏化工,30(3)报道,甲苯咪唑粗品用5倍体积的85%甲酸升温溶解,加入0.4%的活性炭脱色,滤液降温至25℃,加入0.4%晶种,再加入10重量的纯水,抽滤,干燥得到c型甲苯咪唑,收率95%。但该方法制备的c晶型质量不稳定,并会产生大量的废酸,处理较困难,且收率低,原料成本偏高。鉴于上述目前甲苯咪唑转型工艺的缺点,开发一种晶型纯度高、收率高、工艺稳定,没有大量废酸产生的甲苯咪唑c晶型制备方法还有待于进一步探索研究。技术实现要素:针对上述路线的种种缺点,本发明的目的在于提供一种工艺稳定,没有大量废酸产生,收率高的甲苯咪唑c晶型制备新方法,具体技术方案如下:一种制备甲苯咪唑c晶型的制备新方法,经过下述化学化学反应方程式进行:上述反应式制备步骤如下:(1)成盐、脱色:在溶剂中,甲苯咪唑粗品(a晶型)与无机酸(m)成盐反应,加入甲苯咪唑粗品质量3%-5%的活性炭,加热至回流,保持回流1小时,滤除活性炭,得到滤液;(2)转型:将上述滤液降温至50-55℃,加入甲苯咪唑c晶型晶种,降低搅拌转速,降温至40-50℃,加入纯水,保温搅拌1-2小时;(3)中和:向步骤(2)转型得到的液体,滴加碱液调ph至6-7,40-50℃搅拌1-2小时,抽滤,滤饼用纯水洗涤,干燥得到甲苯咪唑c晶型。其中,制备步骤(1)中,无机酸(m)为硝酸、硫酸中的至少一种,用量为甲苯咪唑粗品摩尔量的0.5~1倍;溶剂为乙醇、甲醇、丙醇、异丙醇、正丁醇、水中的至少一种,溶剂的用量为甲苯咪唑粗品重量的2~5倍。制备步骤(2)中,甲苯咪唑c晶型晶种用量为甲苯咪唑粗品重量的0.5%~2%;搅拌转速为35-50转/分钟;加入纯水量为甲苯咪唑粗品重量的3~5倍,加纯水时温度控制40-50℃。制备步骤(3)中所述的碱液为氨水、氢氧化钾水溶液、氢氧化钠水溶液、氢氧化锂水溶液中的至少一种,碱液浓度为10%-50%,滴加时间为1-2小时。本发明的有益效果在于:1、本发明提供了一种甲苯咪唑c晶型的制备新方法,c晶型含量达到99.5%以上。2、本发明避免使用价格高昂的甲酸作为溶剂,不会产生大量难处理的甲酸废水,降低了原料成本和三废处理成本。3、本发明提供了一种甲苯咪唑c晶型的制备新方法收率高,收率达到98.0%以上,且工艺稳定。附图说明图1为甲苯咪唑c晶型红外光谱(ir)谱图。图2为甲苯咪唑c晶型x-衍射谱图。具体实施方式下述通过具体的实施例,对本发明做详细的描述,以下实施例用于解释本发明,而不是对本发明的限制。实施例1一种制备甲苯咪唑c晶型的制备新方法:(1)成盐、脱色:在1l带有温度计和搅拌装置的四口烧瓶中,开启搅拌,加入125g的甲苯咪唑粗品,3倍甲苯咪唑粗品重量比的乙醇和0.3倍的纯水,0.8倍甲苯咪唑粗品摩尔量的硝酸(65%),升温至50℃,加入活性炭4g,升温至回流,保温搅拌1小时。热过滤至2000ml四口瓶中得到滤液。(2)转型:将上述滤液降温至50℃,加入甲苯咪唑c晶型晶种1.0g,拌转速至50转/分,降温至45℃,加入3倍甲苯咪唑粗品重量比的纯水,45℃保温搅拌1小时。(3)中和:滴加10%氨水调ph=6-7,滴加时间控制在1小时左右。保温45℃搅拌1小时,抽滤,滤饼用纯水洗涤,95-100℃下干燥,得甲苯咪唑c晶型123.1g。检测数据如下:ir(kbr):3403.8cm–1,1717.3cm–1,1422.6cm–1,1433.5cm–1,1453.1cm–1;经红外检测,c晶型含量为99.5%x-衍射数据如下:2θ角相对峰高(%)2θ角相对峰高(%)2θ角相对峰高(%)4.920100.018.14814.629.22513.57.7011.319.28713.831.4752.411.8423.819.79739.433.5652.512.33610.521.56012.634.9362.913.3232.323.8034.935.8072.314.5913.524.79021.037.7221.814.8282.425.2899.616.25022.126.83833.8实施例2一种制备甲苯咪唑c晶型的制备新方法:(1)成盐、脱色:在1l带有温度计和搅拌装置的四口烧瓶中,开启搅拌,加入125g的甲苯咪唑粗品,4倍甲苯咪唑粗品重量比的加醇和0.4倍的纯水,0.9倍甲苯咪唑粗品摩尔量的硝酸(65%),升温至55℃,加入活性炭4g,升温至回流,保温搅拌1小时。热过滤至2000ml四口瓶中得到滤液。(2)转型:将上述滤液降温至51℃,加入甲苯咪唑c晶型晶种1.3g,降低搅拌转速至40转/分,降温至45℃,加入3倍甲苯咪唑粗品重量比的纯水,45℃保温搅拌1小时。(3)中和:滴加30%氢氧化钠水溶液调ph=6-7,滴加时间控制在1小时左右。保温45℃搅拌1.5小时,抽滤,滤饼用纯水洗涤,100-105℃下干燥,得甲苯咪唑c晶型123.5g。经红外检测,c晶型含量为99.8%。实施例3一种制备甲苯咪唑c晶型的制备新方法:(1)成盐、脱色:在1l带有温度计和搅拌装置的四口烧瓶中,开启搅拌,加入125g的甲苯咪唑粗品,5倍甲苯咪唑粗品重量比的异丙醇和0.5倍的纯水,0.9倍甲苯咪唑粗品摩尔量的硝酸(65%),加入活性炭4g,升温至回流,保温搅拌1小时。热过滤至2000ml四口瓶中得到滤液。(2)转型:将上述滤液降温至52℃,加入甲苯咪唑c晶型晶种2.5g,降低搅拌转速至35转/分,降温至40℃,加入5倍甲苯咪唑粗品重量比的纯水,40℃保温搅拌1小时。(3)中和:滴加15%氨水调ph=6-7,滴加时间控制在1.5小时左右。保温40℃搅拌1小时,抽滤,滤饼用纯水洗涤,100-105℃下干燥,得甲苯咪唑c晶型123.8g。经红外检测,c晶型含量为99.6%。实施例4一种制备甲苯咪唑c晶型的制备新方法:(1)成盐、脱色:在1l带有温度计和搅拌装置的四口烧瓶中,开启搅拌,加入125g的甲苯咪唑粗品,3倍甲苯咪唑粗品重量比的丙醇和0.3倍的纯水,0.8倍甲苯咪唑粗品摩尔量的硝酸(65%),加入活性炭4g,升温至回流,保温搅拌1小时。热过滤至2000ml四口瓶中得到滤液。(2)转型:将上述滤液降温至50℃,加入甲苯咪唑c晶型晶种2.2g,降低搅拌转速至40转/分,降温至47℃,加入4倍甲苯咪唑粗品重量比的纯水,47℃保温搅拌1小时。(3)中和:滴加20%氨水调ph=6-7,滴加时间控制在1小时左右。保温47℃搅拌2小时,抽滤,滤饼用纯水洗涤,95-100℃下干燥,得甲苯咪唑c晶型123.2g。经红外检测,c晶型含量为100.0%。实施例5一种制备甲苯咪唑c晶型的制备新方法:(1)成盐、脱色:在1l带有温度计和搅拌装置的四口烧瓶中,开启搅拌,加入125g的甲苯咪唑粗品,3.5倍甲苯咪唑粗品重量比的回收乙醇(含量90%),1.0倍甲苯咪唑粗品摩尔量的硝酸(65%),加入活性炭4g,升温至回流,保温搅拌1小时。热过滤至2000ml四口瓶中得到滤液。(2)转型:将上述滤液降温至55℃,加入甲苯咪唑c晶型晶种2.0g,降低搅拌转速至45转/分,降温至43℃,加入5倍甲苯咪唑粗品重量比的纯水,43℃保温搅拌1小时。(3)中和:滴加15%氨水调ph=6-7,滴加时间控制在2小时左右。保温43℃搅拌1.5小时,抽滤,滤饼用纯水洗涤,95-100℃下干燥,得甲苯咪唑c晶型124.0g。经红外检测,c晶型含量为99.8%。实施例6一种制备甲苯咪唑c晶型的制备新方法:(1)成盐、脱色:在1l带有温度计和搅拌装置的四口烧瓶中,开启搅拌,加入125g的甲苯咪唑粗品,3.5倍甲苯咪唑粗品重量比的回收乙醇(含量90%),0.5倍甲苯咪唑粗品摩尔量的硫酸(98%),加入活性炭4g,升温至回流,保温搅拌1小时。热过滤至2000ml四口瓶中得到滤液。(2)转型:将上述滤液降温至55℃,加入甲苯咪唑c晶型晶种2.0g,降低搅拌转速至45转/分,降温至43℃,加入5倍甲苯咪唑粗品重量比的纯水,43℃保温搅拌1小时。(3)中和:滴加50%氢氧化钾水溶液调ph=6-7,滴加时间控制在2小时左右。保温43℃搅拌1.5小时,抽滤,滤饼用纯水洗涤,95-100℃下干燥,得甲苯咪唑c晶型123.9g。经红外检测,c晶型含量为99.8%。本发明不限于上述实施例,凡依据本发明的技术实质对上述实施例作的任何简单、等同变化或修饰,均属于本发明技术范围内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1