制备L-丙交酯和ε-己内酯无规共聚物的方法与流程
本发明涉及聚合物的制备技术领域,尤其涉及一种制备l-丙交酯和ε-己内酯高度无规共聚物的方法。
背景技术:
聚l-丙交酯(plla)和聚ε-己内酯(pcl)的一些重要性质在医学和药学应用中受到广泛重视。同时,它们在一些机械性能在功能上是互补的。plla对大部分药物不具有渗透性,在生物体内的半衰期仅有几周。pcl对一些低分子量的药物都有很好的渗透性,但是它在生物体内半衰期长达一年。而l-丙交酯和ε-己内酯的共聚物可以将二者有机结合起来,达到优缺点互补的效果,获得具有良好渗透效果以及较短降解时间的高分子材料,更好地满足工业需求。目前,将各种催化剂用于催化l-丙交酯和ε-己内酯的共聚反应得到嵌段共聚物和梯度共聚物的例子有很多,但是在常温下得到一种高度无规的共聚物的催化剂几乎没有报道。相关技术主要如下:
2010年,nomura等人选用偕二甲基桥联salen铝苄氧基配合物作为催化剂,90℃下甲苯中催化丙交酯和己内酯共聚,反应10h后两种单体接近完全转化,得到了无规共聚物,但是该体系催化剂用量较高,需要2~3mol%,温度也偏高(参见:n.nomura,a.akita,r.ishiiandm.mizuno,j.am.chem.soc.2010,132,1750–1751)。
2012年,pellecchia等人选用不对称双胺基铝甲基配合物作为催化剂,70℃下甲苯中反应4天两种单体均转化约一半,得到了无规共聚物。虽然反应温度有所降低,但是催化剂反应活性过低(参见:g.li,m.lamberti,d.pappalardoandc.pellecchia,macromolecules2012,45,8614-8620)。
同年,马海燕等人选用不同取代基的胺基桥联双芳氧基双铝甲基配合物作为催化剂,通过升高温度调节l-丙交酯和ε-己内酯的竞聚率。130℃时两种单体竞聚率相当,得到无规聚合物。但是,在这个体系中,要得到无规共聚物所需温度太高,一旦降低温度,则只能得到嵌段、渐变和梯度共聚物(参见:y.wang,h.ma,chem.commun.2012,48,6729–6731)。2016年,该等人选用大位阻单核、双核铝配合物作为催化剂,甲苯中高温下可以得到无规共聚物。该体系所需反应温度高达110℃成为了该催化剂的最大缺点(参见:c.kan,h.ma,rscadv.2016,6,47402–47409)。
2014年,崔冬梅等人选用间苯二胺桥联salan和salen型铝甲基配合物作为催化剂,催化l-丙交酯和ε-己内酯的共聚,得到十分接近无规共聚物的产物,但是催化剂活性偏低(参见:l.li,b.liu,d.liu,c.wu,s.li,b.liu,d.cu,organometallics2014,33,6474-6480)。
2017年,visseaux等人选用混配的烯丙基稀土金属配合物为催化剂,70℃下甲苯中可以得到无规共聚物,但是在反应体系中需要加入链转移试剂,且反应时间长达24h(参见:s.fadlallah,j.jothieswaran,f.capet,f.bonnet,m.visseaux,chem.eur.j.2017,23,15644–15654)。
2018年,姚英明等人选用乙二胺基桥联双芳氧基稀土金属配合物作为催化剂,90℃下反应24h通过酯交换进程得到l-丙交酯和ε-己内酯的无规共聚物,无规化程度(%):l-la-ε-cl链节:ε-cl-ε-cl链节=74:26。但是,该反应必须分步法进行,若采用“一锅煮”的方法或者降低反应温度则只得到嵌段共聚物(参见:h.ouyang,k.nie,d.yuan,y.zhang,d.cui,y.yao,sci.chinachem.2018,61,708-714.)。
技术实现要素:
为解决上述技术问题,本发明的目的是提供一种制备l-丙交酯和ε-己内酯高度无规共聚物的方法,该方法为“一锅煮”方法,选用的催化剂催化活性高,能够在非常温和的条件下催化l-丙交酯和ε-己内酯的共聚反应,所得共聚物为高度无规共聚物。
本发明提供了一种制备l-丙交酯和ε-己内酯无规共聚物的方法,包括以下步骤:
以l-丙交酯和ε-己内酯为原料,以席夫碱桥联的双芳氧基配体稳定的稀土金属配合物为催化剂,30-60℃在有机溶剂中发生开环共聚反应,得到l-丙交酯和ε-己内酯无规共聚物;所述席夫碱桥联的双芳氧基配体稳定的稀土金属配合物与l-丙交酯和ε-己内酯的摩尔比为1:120-500:120-500;所述席夫碱桥联的双芳氧基配体稳定的稀土金属配合物结构式如式(1)和/或式(2)所示:
其中thf为四氢呋喃分子,r为2,6-二叔丁基对甲苯基(c6h2-2,6-tbu2-4-ch3)、叔丁基(tbu)、异丙基(ipr)、苄基(bn)和乙基(et)的一种。
进一步地,上述l-丙交酯和ε-己内酯无规共聚物的制备方法具体包括以下步骤:
(1)将席夫碱桥联的双芳氧基配体稳定的稀土金属配合物溶于有机溶剂,得到催化剂溶液;将单体l-丙交酯和ε-己内酯用有机溶剂溶解,向其中一次性加入上述催化剂溶液,在密闭条件下于30-60℃下反应;
(2)加入终止剂终止反应,得到l-丙交酯和ε-己内酯无规共聚物。
步骤(2)之后,用工业酒精沉降聚合物,过滤洗涤数次,真空干燥箱抽去溶剂得到纯净干燥的产物。
l-丙交酯(l-la)和ε-己内酯(ε-cl)的结构式分别如下:
进一步地,反应时间为2-7h。优选地,反应时间为2-3h。
进一步地,所述催化剂和有机溶剂的比例为2-5mmol:1ml。优选地,所述催化剂和有机溶剂的比例为5mmol:1ml。
进一步地,催化剂、l-la和ε-cl的摩尔比为1:120-500:120-500。优选地,催化剂、l-丙交酯和ε-己内酯的摩尔比为1:120-300:120-300。
进一步地,有机溶剂为甲苯、正己烷或四氢呋喃中的一种。
进一步地,l-la和ε-cl无规共聚物中,l-丙交酯的含量为9-90%。
进一步地,l-la和ε-cl无规共聚物中,ε-己内酯的含量为10-91%。
l-la和ε-cl无规共聚物中,l-la和ε-cl的含量由反应开始时催化剂、l-丙交酯和ε-己内酯的摩尔比、反应时间、反应温度以及各物质的加料顺序所共同决定的。
进一步地,还包括加入终止剂终止聚合反应的步骤。
进一步地,终止剂为乙醇的盐酸溶液或95%乙醇水溶液。
本发明中,席夫碱桥联的双芳氧基配体稳定的稀土金属化合物的化学通式为[lal(or)(thf)n1]n2,l代表桥联席夫碱配体,l=nh[ch2ch2n=ch(2-o-3,5-tbu2c6h2)]2;thf为四氢呋喃;r为2,6-二叔丁基对甲苯基、叔丁基、异丙基、苄基或乙基-;n1代表四氢呋喃个数,n1=0或1;n2=1或2。
本发明利用上述催化剂催化单体l-la与ε-cl的开环共聚,其原理如下:
在聚合引发阶段,相同条件下l-la和ε-cl这两种单体的开环聚合的竞聚率相当(rla≈rcl≈1)。通过对反应进程的跟踪可以证明:在共聚反应中l-la和ε-cl几乎同时开始反应,甚至在前7min,ε-cl的转化率都稍大于l-la;随着反应的进行,l-la的转化速率超过ε-cl,并且伴随酯交换反应的出现,直至反应结束。通过1hnmr和13cnmr谱图追踪,证实形成高度无规共聚物是两种单体近似相等的竞聚率以及酯交换反应共同作用的结果。
借由上述方案,本发明至少具有以下优点:
1)本发明使用的催化剂结构明确,合成方法简单,产率高,分离纯化简单;
2)本发明选用的催化剂活性高,催化剂用量小,其催化剂的用量为反应物l-la/ε-cl的0.2-0.5mol%,共聚物收率高。当催化剂摩尔用量降至0.2mol%,仍能保持较高的催化活性,较少的催化剂用量为产物的提纯提供了方便;
3)本发明公开的制备方法中原料易得;反应条件十分温和,甚至可以在常温下得到高度无规共聚物;反应时间短,不同结构的共聚物可以通过改变两种单体的摩尔比实现,反应操作和后处理过程简单,为实现大批量工业化生产提供可能。
上述说明仅是本发明技术方案的概述,为了能够更清楚了解本发明的技术手段,并可依照说明书的内容予以实施,以下以本发明的较佳实施例并配合附图详细说明如后。
附图说明
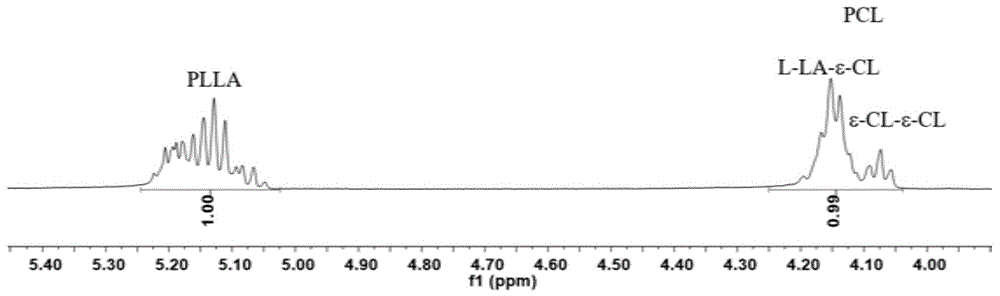
图1是实施例3中所制备的l-丙交酯和ε-己内酯高度无规共聚物在4-5.5ppm区间的核磁氢谱图;
图2是实施例3中所制备的l-丙交酯和ε-己内酯高度无规共聚物在169-174ppm区间的核磁氢谱图。
具体实施方式
下面结合附图和实施例,对本发明的具体实施方式作进一步详细描述。以下实施例用于说明本发明,但不用来限制本发明的范围。
下面结合附图和实施例,对本发明的具体实施方式作进一步详细描述。以下实施例用于说明本发明,但不用来限制本发明的范围。
实施例1lal(oc6h2-2,6-tbu2-4-ch3)(thf)催化l-la和ε-cl(摩尔比50:50)共聚合成高度无规共聚物
手套箱高纯氮保护下,向聚合耐压瓶中加入l-la(0.2394g,1.66mmol)和ε-cl(0.1896g,1.66mmol),用729μl甲苯溶解。称取r=c6h2-2,6-tbu2-4-ch3的金属镧配合物(10.6mg)溶于1000μl的甲苯中,取755μl(0.0083mmol)的催化剂溶液加入到聚合瓶中。在60℃下反应3h后转移出手套箱,取出60μl反应液进行原位1hnmr表征,而后加入95%的乙醇溶液终止反应,用工业酒精沉降,过滤并多次洗涤聚合物,真空干燥12h,得到l-丙交酯和ε-己内酯高度无规共聚物。
从原位核磁图中看出,l-la和ε-cl转化完全;无规化程度(%):l-la-ε-cl链节:ε-cl-ε-cl链节=81:19。对所得固体产物进行gpc分析,测得共聚物的mn=18.0×103g/mol,分子量分布pdi=1.99。
实施例2lal(oc6h2-2,6-tbu2-4-ch3)(thf)催化l-la和ε-cl(摩尔比50:50)共聚合成高度无规共聚物
手套箱高纯氮保护下,向聚合耐压瓶中加入l-la(0.2263g,1.57mmol)和ε-cl(0.1792g,1.57mmol),用688μl甲苯溶解。称取r=c6h2-2,6-tbu2-4-ch3的金属镧配合物(10.6mg)溶于1000μl的甲苯中,取714μl(0.0078mmol)的催化剂溶液加入到聚合瓶中。在30℃下反应3h后转移出手套箱,取出60μl反应液进行原位1hnmr表征,而后加入95%的乙醇溶液终止反应,用工业酒精沉降,过滤并多次洗涤聚合物,真空干燥12h,得到l-丙交酯和ε-己内酯高度无规共聚物。
从原位核磁图中看出,l-la和ε-cl转化完全;无规化程度(%):l-la-ε-cl链节:ε-cl-ε-cl链节=78:22。对所得固体产物进行gpc分析,测得共聚物的mn=26.0×103g/mol,分子量分布pdi=2.23。
实施例3lal(otbu)(thf)催化l-la和ε-cl(摩尔比50:50)共聚合成高度无规共聚物
手套箱高纯氮保护下,向聚合耐压瓶中加入l-la(0.2475g,1.72mmol)和ε-cl(0.1960g,1.72mmol),用805μl甲苯溶解。称取r=tbu的金属镧配合物(9.6mg)溶于1000μl的甲苯中,取729μl(0.0086mmol)的催化剂溶液加入到聚合瓶中。在60℃下反应3h后转移出手套箱,取出60μl反应液进行原位1hnmr表征,而后加入95%的乙醇溶液终止反应,用工业酒精沉降,过滤并多次洗涤聚合物,真空干燥12h,得到l-丙交酯和ε-己内酯高度无规共聚物。
从原位核磁图中看出,l-la和ε-cl转化完全;无规化程度(%):l-la-ε-cl链节:ε-cl-ε-cl链节=80:20。对所得固体产物进行gpc分析,测得共聚物的mn=34.0×103g/mol,分子量分布pdi=2.06。
实施例4lal(otbu)(thf)催化l-la和ε-cl(摩尔比50:50)共聚合成高度无规共聚物
手套箱高纯氮保护下,向聚合耐压瓶中加入l-la(0.2139g,1.48mmol)和ε-cl(0.1694g,1.48mmol),用687μl甲苯溶解。称取r=tbu的金属镧配合物(9.5mg)溶于1000μl的甲苯中,取639μl(0.0074mmol)的催化剂溶液加入到聚合瓶中。在30℃下反应3h后转移出手套箱,取出60μl反应液进行原位1hnmr表征,而后加入95%的乙醇溶液终止反应,用工业酒精沉降,过滤并多次洗涤聚合物,真空干燥12h,得到l-丙交酯和ε-己内酯高度无规共聚物。
从原位核磁图中看出,l-la和ε-cl转化完全;无规化程度(%):l-la-ε-cl链节:ε-cl-ε-cl链节=76:24。对所得固体产物进行gpc分析,测得共聚物的mn=23.1×103g/mol,分子量分布pdi=2.03。
实施例5[lal(oipr)]2催化l-la和ε-cl(摩尔比50:50)共聚合成高度无规共聚物
手套箱高纯氮保护下,向聚合耐压瓶中加入l-la(0.2162g,1.50mmol)和ε-cl(0.1712g,1.50mmol),用556μl甲苯溶解。称取r=ipr的金属镧配合物(7.0mg)溶于1000μl的甲苯中,取784μl(0.0075mmol)的催化剂溶液加入到聚合瓶中。在60℃下反应3h后转移出手套箱,取出60μl反应液进行原位1hnmr表征,而后加入95%的乙醇溶液终止反应,用工业酒精沉降,过滤并多次洗涤聚合物,真空干燥12h,得到l-丙交酯和ε-己内酯高度无规共聚物。
从原位核磁图(图1-2)中看出,原料无剩余,l-la和ε-cl转化完全;无规化程度(%):l-la-ε-cl链节:ε-cl-ε-cl链节=80:20。共聚物玻璃化转变温度:-6.4℃,平均链段长度:lla=0.84;lcl=1.15;对所得固体产物进行gpc分析,测得共聚物的mn=32.9×103g/mol,分子量分布pdi=1.96。
实施例6[lal(oipr)]2催化l-la和ε-cl(摩尔比40:60)共聚合成高度无规共聚物
取l-la(0.1730g,1.20mmol)和ε-己内酯(0.2055g,1.80mmol),按照实施例3的方法进行聚合反应,得到l-la和ε-cl高度无规共聚物。对本实施例制备的产物进行表征,结果如下:
l-la和ε-cl转化完全;无规化程度(%):l-la-ε-cl链节:ε-cl-ε-cl链节=67:33;共聚物玻璃化转变温度:-19.6℃,平均链段长度:lla=0.59;lcl=1.31;对所得固体产物进行gpc分析,测得共聚物的mn=26.9×103g/mol,分子量分布pdi=2.05。
实施例7[lal(oipr)]2催化l-la和ε-cl(摩尔比60:40)共聚合成高度无规共聚物
取l-la(0.2594g,1.80mmol)和ε-己内酯(0.1370g,1.20mmol),按照实施例3的方法进行聚合反应,得到l-la和ε-cl高度无规共聚物。对本实施例制备的产物进行表征,结果如下:
l-la和ε-cl转化完全;无规化程度(%):l-la-ε-cl链节:ε-cl-ε-cl链节=86:14;共聚物玻璃化转变温度:7.9℃,平均链段长度:lla=1.11;lcl=1.11;对所得固体产物进行gpc分析,测得共聚物的mn=30.7×103g/mol,分子量分布pdi=2.01。
实施例8[lal(oipr)]2催化l-la和ε-cl(摩尔比70:30)共聚合成高度无规共聚物
取l-la(0.3027g,2.10mmol)和ε-己内酯(0.1027g,0.90mmol),按照实施例3的方法进行聚合反应,得到l-la和ε-cl高度无规共聚物。对本实施例制备的产物进行表征,结果如下:
l-la和ε-cl转化完全;无规化程度(%):l-la-ε-cl链节:ε-cl-ε-cl链节=89:11;共聚物玻璃化转变温度:19.0℃,平均链段长度:lla=1.89;lcl=1.02;对所得固体产物进行gpc分析,测得共聚物的mn=37.5×103g/mol,分子量分布pdi=1.72。
实施例9[lal(oipr)]2催化l-la和ε-cl(摩尔比50:50)共聚合成高度无规共聚物
手套箱高纯氮保护下,向聚合耐压瓶中加入l-la(0.2274g,1.58mmol)和ε-cl(0.1801g,1.58mmol),用549μl甲苯溶解。称取r=ipr的金属镧配合物(6.7mg)溶于1000μl的甲苯中,取861μl(0.0079mmol)的催化剂溶液加入到聚合瓶中。在30℃下反应3h后转移出手套箱,加入95%的乙醇溶液终止反应,用工业酒精沉降,过滤并多次洗涤聚合物,真空干燥12h,得到l-丙交酯和ε-己内酯高度无规共聚物。
对产物进行表征,l-la的转化率为88%,ε-cl的转化率为94%;无规化程度(%):l-la-ε-cl链节:ε-cl-ε-cl链节=77:23。
实施例10[lal(obn)]2催化l-la和ε-cl(摩尔比50:50)共聚合成高度无规共聚物
手套箱高纯氮保护下,向聚合耐压瓶中加入l-la(0.2291g,1.59mmol)和ε-cl(0.1814g,1.59mmol),用593μl甲苯溶解。称取r=bn的金属镧配合物(7.5mg)溶于1000μl的甲苯中,取827μl(0.0080mmol)的催化剂溶液加入到聚合瓶中。在60℃下反应3h后转移出手套箱,取出60μl反应液进行原位1hnmr表征,而后加入95%的乙醇溶液终止反应,用工业酒精沉降,过滤并多次洗涤聚合物,真空干燥12h,得到l-丙交酯和ε-己内酯高度无规共聚物。
从原位核磁图中看出,l-la和ε-cl转化完全;无规化程度(%):l-la-ε-cl链节:ε-cl-ε-cl链节=80:20。对所得固体产物进行gpc分析,测得共聚物的mn=17.7×103g/mol,分子量分布pdi=1.81。
实施例11[lal(obn)]2催化l-la和ε-cl(摩尔比50:50)共聚合成高度无规共聚物
手套箱高纯氮保护下,向聚合耐压瓶中加入l-la(0.2899g,2.01mmol)和ε-cl(0.2296g,2.01mmol),用953μl甲苯溶解。称取r=bn的金属镧配合物(9.3mg)溶于1000μl的甲苯中,取843μl(0.0100mmol)的催化剂溶液加入到聚合瓶中。在30℃下反应3h后转移出手套箱,取出60μl反应液进行原位1hnmr表征,而后加入95%的乙醇溶液终止反应,用工业酒精沉降,过滤并多次洗涤聚合物,真空干燥12h,得到l-丙交酯和ε-己内酯高度无规共聚物。
从原位核磁图中看出,l-la和ε-cl转化完全;无规化程度(%):l-la-ε-cl链节:ε-cl-ε-cl链节=75:25。对所得固体产物进行gpc分析,测得共聚物的mn=19.4×103g/mol,分子量分布pdi=1.86。
实施例12[lal(oet)]2催化l-la和ε-cl(摩尔比50:50)共聚合成高度无规共聚物
手套箱高纯氮保护下,向聚合耐压瓶中加入l-la(0.2061g,1.43mmol)和ε-cl(0.1632g,1.43mmol),用412μl甲苯溶解。称取r=et的金属镧配合物(5.9mg)溶于1000μl的甲苯中,取865μl(0.0071mmol)的催化剂溶液加入到聚合瓶中。在60℃下反应3h后转移出手套箱,取出60μl反应液进行原位1hnmr表征,而后加入95%的乙醇溶液终止反应,用工业酒精沉降,过滤并多次洗涤聚合物,真空干燥12h,得到l-丙交酯和ε-己内酯高度无规共聚物。
从原位核磁图中看出,l-la和ε-cl转化完全;无规化程度(%):l-la-ε-cl链节:ε-cl-ε-cl链节=83:17。对所得固体产物进行gpc分析,测得共聚物的mn=19.9×103g/mol,分子量分布pdi=1.87。
实施例13[lal(oet)]2催化l-la和ε-cl(摩尔比50:50)共聚合成高度无规共聚物
手套箱高纯氮保护下,向聚合耐压瓶中加入l-la(0.2532g,1.76mmol)和ε-cl(0.2005g,1.76mmol),用932μl甲苯溶解。称取r=et的金属镧配合物(9.9mg)溶于1000μl的甲苯中,取637μl(0.0088mmol)的催化剂溶液加入到聚合瓶中。在30℃下反应3h后转移出手套箱,取出60μl反应液进行原位1hnmr表征,而后加入95%的乙醇溶液终止反应,用工业酒精沉降,过滤并多次洗涤聚合物,真空干燥12h,得到l-丙交酯和ε-己内酯高度无规共聚物。
从原位核磁图中看出,l-la和ε-cl转化完全;无规化程度(%):l-la-ε-cl链节:ε-cl-ε-cl链节=73:27。对所得固体产物进行gpc分析,测得共聚物的mn=20.7×103g/mol,分子量分布pdi=1.89。
以上所述仅是本发明的优选实施方式,并不用于限制本发明,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和变型,这些改进和变型也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!