一种高纯度医药中间体的制备方法与流程
本发明属于医药化工技术领域,更具体地说,涉及一种高纯度医药中间体的制备方法。
背景技术:
作为Schiff碱类化合物中的一种,吡喃酮衍生物在自然界中分布广泛,是许多天然产物分子或医药药物合成的中间体或基本骨架,其具备众多生理活性,对多种生理过程发挥着重要作用,比如止痛活性、抗真菌活性、抗病毒活性、抗癌活性、抵御其它有机体入侵的特性以及利尿活性等。基于上述特性,吡喃酮衍生物在生物医药中是很重要的生物合成中间体。而吡喃酮衍生物中含有吡喃酮结构的4-羟基-6-甲基-[1-(苯基亚氨基)乙基]-2H-吡喃-2-酮衍生物也是一类重要的应用于药理学和生物活性方面的医药中间体。
Schiff碱类化合物的制备往往是通过缩合、加成、重排、消去、偶合等一系列步骤组成的,而一般意义上的碱类化合物通常是由醛或酮与伯胺缩合生成的产物。随着科学技术的进步,人们开始根据实际需要选择不同的反应原料,以达到不同的目的。比如,近年来,研究者开始以脱氢乙酸与胺类及其衍生物作为反应原料,通过发生偶合反应而制备得到4-羟基-6-甲基-[1-(苯基亚氨基)乙基]-2H-吡喃-2-酮衍生物,制备过程中往往需要有机碱或酸作为催化剂。
Antonio J.Demuner等以脱氢乙酸(α,γ-二乙酰基乙酰乙酸)和芳香胺作为反应原料,以三乙胺作为反应催化剂、1,4-二氧环己烷作为反应溶剂的条件下合成了一系列的4-羟基-6-甲基-[1-(苯基亚氨基)乙基]-2H-吡喃-2-酮衍生物(Preparation of achiral and chiral(E)-enaminopyran-2,4-diones and their phytotoxic activity[J],Journal of Agricultural and Food Chemistry,2009,57:1399~1405)。但是该方法存在以下问题:一是制备过程反应时间长,长达16h;二是制备得到的产物纯度低,需要进一步的提纯操作;三是反应催化剂及溶剂无法循环使用,在实际应用中多受限制。
随后,臧洪俊等仍以脱氢乙酸以与芳香胺为原料,但是,采用酸性离子液体[BMIM-SO3H]HSO4作为催化剂,并在40℃水浴下采用超声辐射辅助,反应终止后,再经旋蒸除去有机溶剂,将得到的残留物用乙醇洗涤,必要时硅胶柱层析提纯产物以获得纯产品,反应制备了一系列的4-羟基-6-甲基-[1-(苯基亚氨基)乙基]-2H-吡喃-2-酮衍生物,大大缩短了反应时间,且产物收率高,催化剂可重复使用(超声波辐射下离子液体催化合成4-羟基-6-甲基-[1-(苯基亚氨基)乙基]-2H-吡喃-2-酮衍生物[J],有机化学,2012,32:2193~2197)。但是该方法仍然存在以下几个问题:一是所得产物仍需进一步处理才能提高纯度;二是能回收再利用的物质有限,只能实现催化剂的回收,且催化剂的回收过程复杂、耗费成本较高;三是目前对于含有咪唑、吡啶等母体结构的酸性离子液体来说,其本身制备成本就比较高,且生物降解性差,此外,随着对离子液体进一步研究还发现,该种类型的离子液体还具有一定的毒性。
因此,开发一种绿色、高效、方便快捷地高纯度医药中间体4-羟基-6-甲基-[1-(苯基亚氨基)乙基]-2H-吡喃-2-酮衍生物的合成方法具有切实的实际意义。
技术实现要素:
1.要解决的问题
针对现有技术中制备4-羟基-6-甲基-[1-(苯基亚氨基)乙基]-2H-吡喃-2-酮衍生物的方法具收率低、反应溶剂无法回用,制备出的产物需借助其他技术手段进行提纯的问题,本发明提供一种高纯度医药中间体4-羟基-6-甲基-[1-(苯基亚氨基)乙基]-2H-吡喃-2-酮衍生物的制备方法。
2.技术方案
为了解决上述问题,本发明所采用的技术方案如下:
一种高纯度医药中间体的制备方法,以脱氢乙酸和芳香胺为原料,在异丙醇水溶液中,经催化剂催化缩合,得到4-羟基-6-甲基-[1-(苯基亚氨基)乙基]-2H-吡喃-2-酮衍生物。反应过程仅有微量副产物生产,反应结束后直接形成高纯度的4-羟基-6-甲基-[1-(苯基亚氨基)乙基]-2H-吡喃-2-酮衍生物,无需进一步借助硅胶柱层析提纯,且产物的收率高;将4-羟基-6-甲基-[1-(苯基亚氨基)乙基]-2H-吡喃-2-酮衍生物分离出来后得到残余滤液,无需进行任何处理,可直接加入原料后进行下一轮缩合反应,能够简单、便捷地实现滤液(异丙醇水溶液溶剂与催化剂)的循环回用。
优选地,所述异丙醇水溶液中异丙醇:水的体积比浓度为46%~55%。
优选地,所述异丙醇水溶液中异丙醇:水的体积比浓度为50%~54%。
优选地,所述异丙醇水溶液中异丙醇:水的体积比浓度为41%~53%。
优选地,所述异丙醇水溶液中异丙醇:水的体积比浓度为52%~53%。
进一步的,所述医药中间体的制备方法,具体步骤如下:将脱氢乙酸、芳香胺以及催化剂加入到异丙醇水溶液中,混匀,加热回流处理50~100min,直接冷却静置即可析出固体产物,将固体产物分离洗涤,真空干燥,即得高纯度的4-羟基-6-甲基-[1-(苯基亚氨基)乙基]-2H-吡喃-2-酮衍生物。无需进一步借助硅胶柱层析或重结晶等其他手段进行提纯,简化了实验步骤。
为了进一步去除杂质,保证产物纯度,利用无水乙醇对分离出来的固体产物进行洗涤。
将固体产物分离后得到滤液,所得滤液可循环使用。反应过程中副产物极少,因此滤液中几乎无残留杂质,可以直接回用,不会因杂质对下一轮反应产生影响;另外,反应过程中溶剂与催化剂的损失几乎可以不计,不会因溶剂量以及催化剂浓度变化对下一轮反应产生影响;并且经多次循环实验验证可以看出,滤液重复且连续回收再利用5或6次后,所制备得到的4-羟基-6-甲基-[1-(苯基亚氨基)乙基]-2H-吡喃-2-酮衍生物纯度仍大于98%,收率仍高于60%。
进一步的,所用催化剂为1,3,6-萘三磺酸。1,3,6-萘三磺酸与异丙醇水溶液共同组成原料进行化学合成的反应-催化溶剂体系,二者产生协同作用,异丙醇水溶液能够改变1,3,6-萘三磺酸的催化方式(给H+方式),减少反应过程中的副产物,而1,3,6-萘三磺酸可以提供足够的活性催化基团,提高产物收率。
原料配比、催化剂种类、催化剂用量、反应溶剂等共同构成了最终的反应体系,而对于任何化学合成或生产来说往往牵一发而动全身,其中任何一方面的改变都会导致后期产物的纯度、收率降低以及反应过程中副产物的生成量增加,进而影响效果,基于此:
优选地,所述芳香胺与脱氢乙酸的添加摩尔比为(1.0~1.2):1。
优选地,异丙醇水溶液以毫升计的体积量为脱氢乙酸以毫摩尔计的物质的量的7~10倍。
优选地,1,3,6-萘三磺酸与脱氢乙酸的添加摩尔比为(0.04~0.07):1。
优选地,上述的1,3,6-萘三磺酸,其结构式如下:
利用1,3,6-萘三磺酸催化脱氢乙酸和芳香胺制备4-羟基-6-甲基-[1-(苯基亚氨基)乙基]-2H-吡喃-2-酮衍生物的反应式如下:
其中芳香胺为4-硝基苯胺、苯甲胺、4-氯苯胺、2-氯苯胺、3-氯苯胺、4-氟苯胺、4-甲基-3-氯苯胺、4-甲基苯胺、4-甲氧基苯胺中的任一种。
3.有益效果
相比于现有技术,本发明的有益效果为:
(1)本发明提供的高纯度医药中间体的制备方法,以异丙醇水溶液为反应溶剂,其可以改变催化剂的催化方式,减少反应过程中生成的副产物,这样一方面能够保证原料到产物的有效生成率,产物收率高,另一方面能够提高产物纯度,无需进一步层析或重结晶提纯;
(2)本发明提供的高纯度医药中间体的制备方法,分离产物后的滤液可以循环回用,绿色环保,且多次循环回用仍能保障产物的收率与纯度,滤液回用时无需做任何处理即可循环使用,回收再利用简单;
(3)本发明提供的高纯度医药中间体的制备方法,以1,3,6-萘三磺酸为催化剂,与异丙醇水溶液共同组成反应催化体系,二者产生协同作用,异丙醇水溶液改变1,3,6-萘三磺酸的催化方式(给H+方式),减少反应过程中的副产物,提高了产物的选择性,在原料用量相同的情况下(以脱氢乙酸为计量基准)能获得更多的产物,整个制备方法经济高效,绿色环保。
附图说明
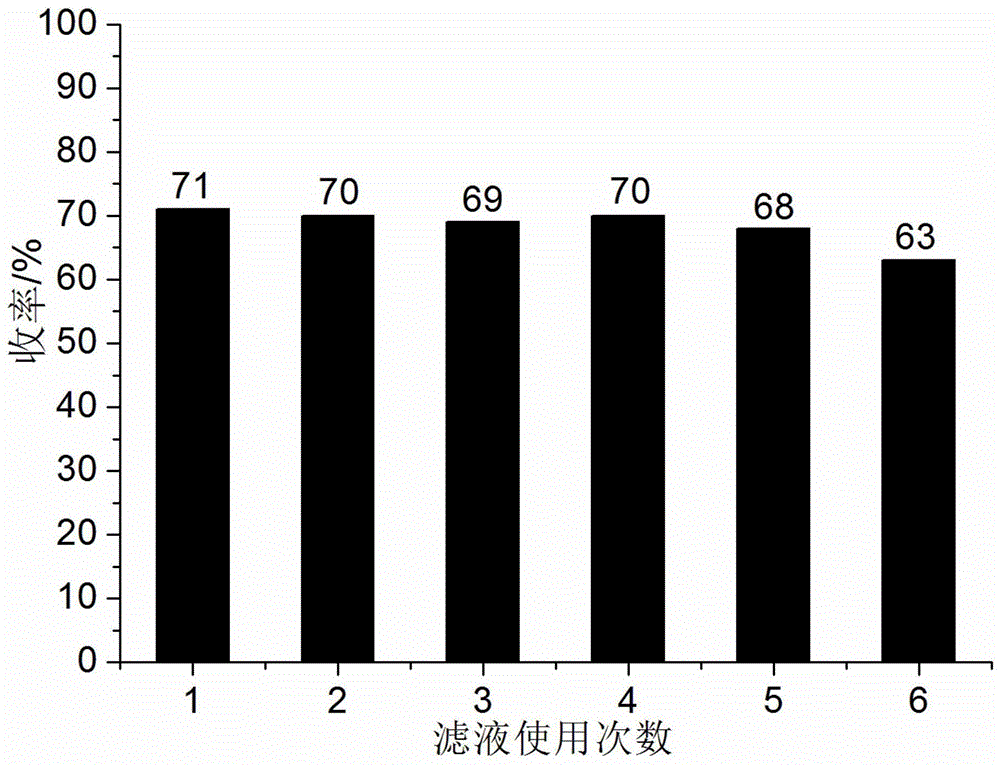
图1为本发明实施例10中1,3,6-萘三磺酸和异丙醇水溶液组成的催化体系在催化制备3-[1-(4-硝基苯基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮反应中循环使用时的产物收率变化图;
图2为本发明实施例11中1,3,6-萘三磺酸和异丙醇水溶液组成的催化体系在催化制备3-[1-(苯甲基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮反应中循环使用时的产物收率变化图;
图3为本发明实施例12中1,3,6-萘三磺酸和异丙醇水溶液组成的催化体系在催化制备3-[1-(3-氯4-甲基苯基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮反应中循环使用时的产物收率变化图。
具体实施方式
本发明的实质特点和显著效果可以从下述的实施例中得以体现,但它们并不对本发明作任何限制,本领域的技术人员根据本发明的内容做出一些非本质的改进和调整,均属于本发明的保护范围。
下面通过具体实施例对本发明作进一步的说明,其中实施例中反应产物的红外光谱测试表征使用的是德国Bruker公司的型号为EQUINOX 55红外光谱仪(KBr压片);氢谱核磁共振表征采用的是德国Bruker公司的型号为AVANCE 300MHz的核磁共振仪;反应产物的熔点采用毛细管法测定。
实施例1
向盛有9ml体积比浓度52%的异丙醇水溶液的带有冷凝管和搅拌磁子的50ml单口瓶中加入1.2mmol 4-硝基苯胺、1.0mmol脱氢乙酸和0.07mmol的1,3,6-萘三磺酸催化剂,室温下磁力搅拌混匀。加热至有回流产生,保持回流反应时长为91min,TLC(薄板层析)检测,原料点消失,反应结束冷却至室温析出大量固体,静置,抽滤,滤渣利用3mL的无水乙醇洗涤3次(3ml×3)、80℃下真空干燥后得到3-[1-(4-硝基苯基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮,高效液相色谱测定其纯度为98.9%,计算收率为72%。滤液中直接加入4-硝基苯胺和脱氢乙酸后进行重复使用。
3-[1-(4-硝基苯基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮:白色固体;m.p.154~156℃;IR(KBr)ν:3396,3033,2925,1698,1647,1562,1468,1379,1066,942,835,762cm-1;1H NMR(300MHz,CDCl3):δ=2.18(s,3H,CH3),2.36(s,3H,CH3),2.55(s,3H,CH3),5.73(s,1H,CH),7.02(d,J=6.5Hz,2H,ArH),7.21(d,J=6.1Hz,2H,ArH),15.64(s,1H,OH)。
实施例2
向盛有7ml体积比浓度46%的异丙醇水溶液的带有冷凝管和搅拌磁子的50ml单口瓶中加入1.0mmol苯甲胺、1.0mmol脱氢乙酸和0.04mmol的1,3,6-萘三磺酸催化剂,室温下磁力搅拌混匀。加热至有回流产生,保持回流反应时长为54min,TLC(薄板层析)检测,原料点消失,反应结束冷却至室温析出大量固体,静置,抽滤,滤渣经无水乙醇洗涤(3ml×3)、80℃下真空干燥后得到3-[1-(苯甲基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮,高效液相色谱测定其纯度为98.6%,计算收率为86%。滤液中直接加入苯甲胺和脱氢乙酸后进行重复使用。
3-[1-(苯甲基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮:淡黄色固体;m.p.79~81℃;IR(KBr)ν:3439,3055,2914,1696,1641,1568,1470,1384,1057,993,872,725cm-1;1H NMR(300MHz,CDCl3):δ=2.14(s,3H,CH3),2.66(s,3H,CH3),4.65(d,J=5.4Hz,2H,CH2),5.69(s,1H,CH),7.24~7.29(m,2H,ArH),7.35(d,J=6.6Hz,1H,ArH),7.37(t,J=7.5Hz,2H,ArH),14.52(s,1H,OH)。
实施例3
向盛有8ml体积比浓度50%的异丙醇水溶液的带有冷凝管和搅拌磁子的50ml单口瓶中加入1.1mmol 4-氯苯胺、1.0mmol脱氢乙酸和0.06mmol的1,3,6-萘三磺酸催化剂,室温下磁力搅拌混匀。加热至有回流产生,保持回流反应时长为79min,TLC(薄板层析)检测,原料点消失,反应结束冷却至室温析出大量固体,静置,抽滤,滤渣经无水乙醇洗涤(3ml×3)、80℃下真空干燥后得到3-[1-(4-氯苯基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮,高效液相色谱测定其纯度为99.1%,计算收率为79%。滤液中直接加入4-氯苯胺和脱氢乙酸后进行重复使用。
3-[1-(4-氯苯基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮:白色固体;m.p.138~140℃;IR(KBr)ν:3401,3077,2723,1714,1649,1563,1462,1326,1088,947,839,716cm-1;1H NMR(300MHz,CDCl3):δ=2.11(s,3H,CH3),2.46(s,3H,CH3),5.75(s,1H,CH),7.39(d,J=8.5Hz,2H,ArH),7.52(d,J=8.5Hz,2H,ArH),15.69(s,1H,OH)
实施例4
向盛有8ml体积比浓度50%的异丙醇水溶液的带有冷凝管和搅拌磁子的50ml单口瓶中加入1.1mmol 2-氯苯胺、1.0mmol脱氢乙酸和0.07mmol的1,3,6-萘三磺酸催化剂,室温下磁力搅拌混匀。加热至有回流产生,保持回流反应时长为84min,TLC(薄板层析)检测,原料点消失,反应结束冷却至室温析出大量固体,静置,抽滤,滤渣经无水乙醇洗涤(3ml×3)、80℃下真空干燥后得到3-[1-(2-氯苯基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮,高效液相色谱测定其纯度为99.2%,计算收率为74%。滤液中直接加入2-氯苯胺和脱氢乙酸后进行重复使用。
3-[1-(2-氯苯基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮:白色固体;m.p.159~161℃;IR(KBr)ν:3430,3079,2925,1696,1644,1567,1459,1362,1060,945,857,754cm-1;1H NMR(300MHz,CDCl3):δ=2.15(s,3H,CH3),2.49(s,3H,CH3),5.77(s,1H,CH),7.23(t,J=7.5Hz,1H,ArH),7.29~7.35(m,2H,ArH),7.55(dd,J1=1.5Hz,J2=7.1Hz,1H,ArH),15.93(s,1H,OH)
实施例5
向盛有9ml体积比浓度52%的异丙醇水溶液的带有冷凝管和搅拌磁子的50ml单口瓶中加入1.1mmol 3-氯苯胺、1.0mmol脱氢乙酸和0.06mmol的1,3,6-萘三磺酸催化剂,室温下磁力搅拌混匀。加热至有回流产生,保持回流反应时长为68min,TLC(薄板层析)检测,原料点消失,反应结束冷却至室温析出大量固体,静置,抽滤,滤渣经无水乙醇洗涤(3ml×3)、80℃下真空干燥后得到3-[1-(3-氯苯基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮,高效液相色谱测定其纯度为98.9%,计算收率为80%。滤液中直接加入3-氯苯胺和脱氢乙酸后进行重复使用。
3-[1-(3-氯苯基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮:白色固体;m.p.116~118℃;IR(KBr)ν:3427,3061,2644,1703,1651,1564,1462,1383,1068,996,837,792cm-1;1H NMR(300MHz,CDCl3):δ=2.13(s,3H,CH3),2.56(s,3H,CH3),5.75(s,1H,CH),7.04(d,J=7.5Hz,1H,ArH),7.18(s,1H,ArH),7.31~7.38(m,2H,ArH),15.90(s,1H,OH)
实施例6
向盛有9ml体积比浓度52%的异丙醇水溶液的带有冷凝管和搅拌磁子的50ml单口瓶中加入1.1mmol 4-氟苯胺、1.0mmol脱氢乙酸和0.06mmol的1,3,6-萘三磺酸催化剂,室温下磁力搅拌混匀。加热至有回流产生,保持回流反应时长为81min,TLC(薄板层析)检测,原料点消失,反应结束冷却至室温析出大量固体,静置,抽滤,滤渣经无水乙醇洗涤(3ml×3)、80℃下真空干燥后得到3-[1-(4-氟苯基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮,高效液相色谱测定其纯度为98.7%,计算收率为76%。滤液中直接加入4-氟苯胺和脱氢乙酸后进行重复使用。
3-[1-(4-氟苯基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮:白色固体;m.p.146~148℃;IR(KBr)ν:3425,3083,2929,1728,1664,1579,1481,1333,1067,1005,836,774cm-1;1H NMR(300MHz,CDCl3):δ=2.14(s,3H,CH3),2.55(s,3H,CH3),5.76(s,1H,CH),7.17(d,J=6.5Hz,4H,ArH),15.73(s,1H,OH)
实施例7
向盛有9ml体积比浓度52%的异丙醇水溶液的带有冷凝管和搅拌磁子的50ml单口瓶中加入1.0mmol 4-甲基-3-氯苯胺、1.0mmol脱氢乙酸和0.05mmol的1,3,6-萘三磺酸催化剂,室温下磁力搅拌混匀。加热至有回流产生,保持回流反应时长为59min,TLC(薄板层析)检测,原料点消失,反应结束冷却至室温析出大量固体,静置,抽滤,滤渣经无水乙醇洗涤(3ml×3)、80℃下真空干燥后得到3-[1-(3-氯4-甲基苯基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮,高效液相色谱测定其纯度为98.4%,计算收率为84%。滤液中直接加入4-甲基-3-氯苯胺和脱氢乙酸后进行重复使用。
3-[1-(3-氯4-甲基苯基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮:浅黄色固体;m.p.167~169℃;IR(KBr)ν:3437,3078,2981,1704,1652,1559,1468,1356,1063,950,832,763cm-1;1H NMR(300MHz,CDCl3):δ=2.12(s,3H,CH3),2.35(s,3H,CH3),2.52(s,3H,CH3),5.70(s,1H,CH),6.93(d,J=8.0Hz,1H,ArH),7.13(s,1H,ArH),7.24(d,J=8.0Hz,1H,ArH),15.74(s,1H,OH)
实施例8
向盛有9ml体积比浓度54%的异丙醇水溶液的带有冷凝管和搅拌磁子的50ml单口瓶中加入1.1mmol 4-甲基苯胺、1.0mmol脱氢乙酸和0.06mmol的1,3,6-萘三磺酸催化剂,室温下磁力搅拌混匀。加热至有回流产生,保持回流反应时长为66min,TLC(薄板层析)检测,原料点消失,反应结束冷却至室温析出大量固体,静置,抽滤,滤渣经无水乙醇洗涤(3ml×3)、80℃下真空干燥后得到3-[1-(4-甲基苯基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮,高效液相色谱测定其纯度为99.0%,计算收率为81%。滤液中直接加入4-甲基苯胺和脱氢乙酸后进行重复使用。
3-[1-(4-甲基苯基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮:白色固体;m.p.154~156℃;IR(KBr)ν:3402,3038,2932,1704,1655,1569,1477,1386,1071,950,841,768cm-1;1H NMR(300MHz,CDCl3):δ=2.15(s,3H,CH3),2.37(s,3H,CH3),2.56(s,3H,CH3),5.71(s,1H,CH),7.01(d,J=6.2Hz,2H,ArH),7.20(d,J=6.1Hz,2H,ArH),15.63(s,1H,OH)
实施例9
向盛有10ml体积比浓度55%的异丙醇水溶液的带有冷凝管和搅拌磁子的50ml单口瓶中加入1.2mmol的4-甲氧基苯胺、1.0mmol脱氢乙酸和0.07mmol的1,3,6-萘三磺酸催化剂,室温下磁力搅拌混匀。加热至有回流产生,保持回流反应时长为87min,TLC(薄板层析)检测,原料点消失,反应结束冷却至室温析出大量固体,静置,抽滤,滤渣经无水乙醇洗涤(3ml×3)、80℃下真空干燥后得到3-[1-(4-甲氧基苯基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮,高效液相色谱测定其纯度为98.8%,计算收率为72%。滤液中直接加入4-甲氧基苯胺和脱氢乙酸后进行重复使用。
4-[1-(4-甲氧基苯基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮:黄色固体;m.p.180~182℃;IR(KBr)ν:3437,3085,2927,1692,1615,1561,1474,1328,1066,942,830,712cm-1;1H NMR(300MHz,CDCl3):δ=2.17(s,3H,CH3),2.54(s,3H,CH3),3.81(s,3H,CH3),5.74(s,1H,CH),6.93(d,J=8.6Hz,2H,ArH),7.08(d,J=8.6Hz,2H,ArH),15.56(s,1H,OH)
实施例10
实施例1反应结束后,滤液未经任何处理按照实施例1中原料的加入量和反应条件进行下一批次制备反应,这样重复进行6次,产物3-[1-(4-硝基苯基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮的液相色谱纯度均大于98.5%,其计算收率变化见图1。
实施例11
实施例2反应结束后,滤液未经任何处理按照实施例2中原料的加入量和反应条件进行下一批次制备反应,这样重复进行5次,产物3-[1-(苯甲基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮的液相色谱纯度均大于98.2%,其计算收率变化见图2。
实施例12
实施例7反应结束后,滤液未经任何处理按照实施例7中原料的加入量和反应条件进行下一批次制备反应,这样重复进行6次,产物3-[1-(3-氯4-甲基苯基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮的液相色谱纯度均大于98.0%,其计算收率变化见图3。
对比例1
向盛有9ml的分析纯的异丙醇的带有冷凝管和搅拌磁子的50ml单口瓶中加入1.2mmol4-硝基苯胺、1.0mmol脱氢乙酸和0.07mmol的1,3,6-萘三磺酸催化剂,室温下磁力搅拌混匀。加热至有回流产生,保持回流反应时长为91min,TLC(薄板层析)检测,经检测发现,合成产物的原料点仍然存在。反应结束冷却至室温后却未有固体析出。然后,经探究发现,产物均溶解于了反应液(滤液)中,且利用高效液相色谱对反应液进行检测发现3-[1-(4-硝基苯基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮的含量仅为43.3%(外标法),经计算收率仅为24%。
对比例2
向盛有9ml体积比浓度52%的乙醇水溶液的带有冷凝管和搅拌磁子的50ml单口瓶中加入1.2mmol 4-硝基苯胺、1.0mmol脱氢乙酸和0.07mmol的1,3,6-萘三磺酸催化剂,室温下磁力搅拌混匀。加热至有回流产生,保持回流反应时长为91min,TLC(薄板层析)检测,原料点没有完全消失,反应结束冷却至室温仅仅析出少量固体,静置,抽滤,滤渣经无水乙醇洗涤(3ml×3)、80℃下真空干燥后得到3-[1-(4-硝基苯基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮,高效液相色谱测定其纯度为92.4%,计算收率仅仅为38%。
对比例3
向盛有9ml体积比浓度26%的异丙醇水溶液的带有冷凝管和搅拌磁子的50ml单口瓶中加入1.2mmol 4-硝基苯胺、1.0mmol脱氢乙酸和0.07mmol的1,3,6-萘三磺酸催化剂,原料无法完全溶解,室温下磁力搅拌混匀。加热至有回流产生,保持回流反应时长为91min,TLC(薄板层析)检测,原料点没有完全消失,反应结束冷却至室温有大量固体,静置,抽滤,滤渣经无水乙醇洗涤(3ml×3)、80℃下真空干燥后得到淡黄色固体,高效液相色谱测定得3-[1-(4-硝基苯基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮的含量仅为78.6%(外标法),计算收率为43%。
对比例4
向盛有9ml体积比浓度78%的异丙醇水溶液的带有冷凝管和搅拌磁子的50ml单口瓶中加入1.2mmol 4-硝基苯胺、1.0mmol脱氢乙酸和0.07mmol的1,3,6-萘三磺酸催化剂,原料完全溶解,室温下磁力搅拌混匀。加热至有回流产生,保持回流反应时长为91min,TLC(薄板层析)检测,原料点没有完全消失,反应结束冷却至室温未有固体析出。反应液经过高效液相色谱测定得3-[1-(4-硝基苯基亚氨基)乙基]-4-羟基-6-甲基-2H-吡喃-2-酮的含量仅为56.9%(外标法),计算收率为35%。
- 还没有人留言评论。精彩留言会获得点赞!