一种噻唑烷酮化合物的合成方法与流程
本发明属于有机合成领域,具体涉及一种噻唑烷酮化合物的合成方法。
背景技术:
噻唑烷酮结构存在于许多天然产物中,在药物以及现代有机合成化学中具有重要作用。噻唑烷酮化合物代表着一类众所周知的专利药物和不同研究阶段的物质,具有降血糖、抗炎、利胆、抗肿瘤、利尿、免疫刺激等活性,还有杀菌、杀虫、抗甲状腺病等作用,因此受到了人们的广泛关注。现有技术中,主流的噻唑烷酮合成方法有如下几种:早期最简单的合成方法是通过氨基硫醇与提供羰基化合物的环化反应合成,或氨基乙醇在蒙脱土或吡啶等催化下直接与氧硫化碳反应合成(tetrahedronlett.1975,16,1969;syn.comm.1987,17,1577)。此类方法中,氨基硫醇市面上鲜有销售,一定程度上限制了该方法的应用。另外也有报道以环酮、巯基乙酸和碳酸铵为原料在苯中回流反应合成产物(org.prep.proc.int.1992,24,81)。由于该方法含有巯基原料,因此通常会发出令人不愉快的气味,并且兼有产率不高等缺陷。也有通过过渡金属催化得到的(j.org.chem.2018,83,1059;angew.chem.int.ed.2015,54,10944;acschem.neurosci.2016,7,897),但是目前的合成方法中钌金属配合物的价格比较昂贵,且后处理过于繁琐影响了工业化效率。因此,亟需找到合成路线更优的噻唑烷酮类化合物的合成方法。
技术实现要素:
本发明的目的在于克服现有制备技术的缺陷,提供一种起始原料简单易得、产物产率高、操作简便的噻唑烷酮类化合物的合成方法。
本发明的技术方案为:一种噻唑烷酮化合物的合成方法,其中,以苄基卤化合物、2-(烷基硫基)-4,5-二氢噻唑为反应原料,以碱试剂和碘为催化剂,在溶剂中加热条件下用一锅法反应得到噻唑烷酮类化合物,反应式如下所示:
其中苄基卤化合物中,r具有以下通式:
其中,r0为具有1-22个碳原子的烷基,当碳原子个数大于3时,所述烷基结构选自于直链、支链或者环状烷基链。
r1-r5为连在苯环上的取代基,独立地选自于氢原子、卤素、烯基、炔基、芳基、羟基、氨基、羰基、氨基、羧基、酯基、氰基、苯基、苄基、硝基、或者具有1-22个碳原子的烷基,当碳原子个数大于3时,所述烷基结构选自于直链、支链或者环状烷基链。
x选自于卤素。
进一步地,x选自于溴原子。
进一步地,所述一种噻唑烷酮化合物的合成方法,其中碱试剂选自碳酸钾、氢氧化钾、醋酸钾、碳酸氢钾、叔丁醇钾中的一种或几种。
进一步地,所述一种噻唑烷酮化合物的合成方法,其中碱试剂为叔丁醇钾。
进一步地,所述一种噻唑烷酮化合物的合成方法,其中溶剂选自碳酸二甲酯、甲苯、四氯化碳、乙醇、四氢呋喃、乙腈、水中的一种。
进一步地,所述一种噻唑烷酮化合物的合成方法,其中溶剂为碳酸二甲酯。
进一步地,所述一种噻唑烷酮化合物的合成方法,其中加热的温度为30-120℃,反应时间为10-24小时。
进一步地,所述一种噻唑烷酮化合物的合成方法,其中加热的温度为80-100℃,反应时间为16-20小时。
进一步地,所述一种噻唑烷酮化合物的合成方法,其中苄基卤化合物、2-(烷基硫基)-4,5-二氢噻唑、碱试剂和碘的摩尔比为1:0.5:0.5:1。
进一步地,所述噻唑烷酮化合物的合成方法,所述r0选自于甲基。
进一步地,所述一种噻唑烷酮化合物的合成方法,其中r1-r5中至少有4个选自于氢原子。
本发明具有以下有益效果:
本发明的以价格低廉的苄基卤和2-(烷基硫基)-4,5-二氢噻唑作为反应原料、碱试剂、碘作为催化剂,在溶剂中加热条件下一锅法反应即可得到噻唑烷酮类化合物。该合成方法起始原料制备简单,操作方便,且最终获得的终产物产率较高;除了终产物外,转化过程中的中间体为2-恶唑烷硫酮,极易转化为最终产物噻唑烷酮,因此无需进行中间体分离。本发明使用的原料量较少,价格低廉,能减少资金和劳动力的投入量,为噻唑烷酮类化合物提供一种简洁高效的制备方法。本发明具有良好的实用价值和社会经济效率,对同类产品及下游产品的工艺开发具有很好的借鉴意义。
附图说明
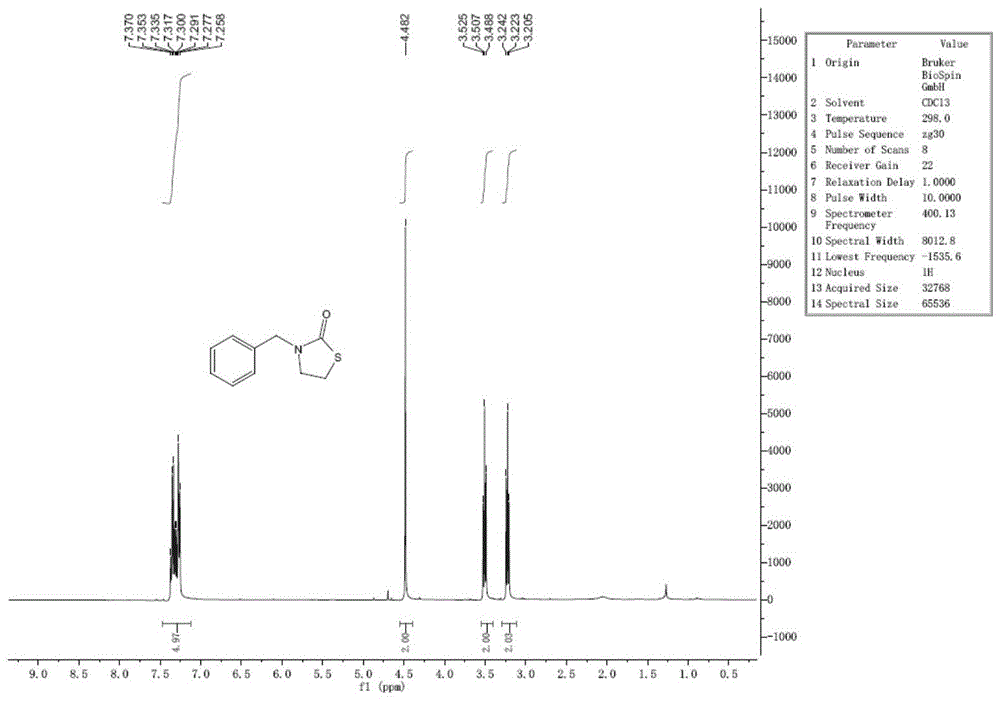
图1为实施例1-1~实施例1-3中3-苄基噻唑烷-2-酮的核磁共振氢谱图。
图2为实施例1-1~实施例1-3中3-苄基噻唑烷-2-酮的核磁共振碳谱图。
图3为实施例2-1~实施例2-3中3-(2-甲基苄基)噻唑烷-2-酮的核磁共振氢谱图。
图4为实施例2-1~实施例2-3中3-(2-甲基苄基)噻唑烷-2-酮的核磁共振碳谱图。
图5为实施例3中3-(4-甲基苄基)噻唑烷-2-酮的核磁共振氢谱图。
图6为实施例3中3-(4-甲基苄基)噻唑烷-2-酮的核磁共振碳谱图。
图7为实施例4中3-(4-(叔丁基)苄基)噻唑烷-2-酮的核磁共振氢谱图。
图8为实施例4中3-(4-(叔丁基)苄基)噻唑烷-2-酮的核磁共振碳谱图。
图9为实施例5中3-(4-溴苄基)噻唑烷-2-酮的核磁共振氢谱图。
图10为实施例5中3-(4-溴苄基)噻唑烷-2-酮的核磁共振碳谱图。
图11为噻唑烷酮化合物的合成化学方程式通式。
具体实施方式
以下结合具体实施例和对本发明中噻唑烷酮化合物合成方法作进一步的说明。但本发明所要求的保护范围并不局限于实施例所涉及的范围。
实施例1-1:3-苄基噻唑烷-2-酮的合成方法(1)
在反应管中,依次加入苄溴(119μl,1mmol)、2-(甲硫基)-4,5-二氢噻唑(56μl,0.5mmol)、氢氧化钾(23mg,0.5mmol)、碘(256mg,1.0mmol)和碳酸二甲酯(1ml),在反应温度为80℃情况下电磁搅拌反应,反应时间为16小时。反应完成后,将溶剂用旋转蒸发去除,混合物用柱层析分离,洗脱剂为乙酸乙酯和石油醚(体积比为1:9),分离后得到淡黄色液体(73mg,76%)。
实施例1-2:3-苄基噻唑烷-2-酮的合成方法(2)
在反应管中,依次加入苄溴(119μl,1mmol)、2-(甲硫基)-4,5-二氢噻唑(56μl,0.5mmol)、碳酸钾(69mg,0.5mmol)、碘(256mg,1.0mmol)和碳酸二甲酯(1ml),在反应温度为80℃情况下电磁搅拌反应,反应时间为16小时。反应完成后,将溶剂用旋转蒸发去除,混合物用柱层析分离,洗脱剂为乙酸乙酯和石油醚(体积比为1:9),分离后得到淡黄色液体(80mg,83%)。
实施例1-3:3-苄基噻唑烷-2-酮的合成方法(3)
在反应管中,依次加入苄溴(119μl,1mmol)、2-(甲硫基)-4,5-二氢噻唑(56μl,0.5mmol)、叔丁醇钾(56mg,0.5mmol)、碘(256mg,1.0mmol)和碳酸二甲酯(1ml),在反应温度为100℃情况下电磁搅拌反应,反应时间为16小时。反应完成后,将溶剂用旋转蒸发去除,混合物用柱层析分离,洗脱剂为乙酸乙酯和石油醚(体积比为1:9),分离后得到淡黄色液体(83mg,86%)。
产物表征数据如下:
1hnmr(400mhz,cdcl3):δ=7.37-7.26(m,5h),4.48(s,2h),3.51(t,2h,j=7.2hz),3.22(t,2h,j=7.2hz)。
13c{1h}nmr(100mhz,cdcl3):δ=172.2,136.0,128.8,128.1,127.9,48.6,48.0,25.5.hrms(ei):c10h12ons[m+h]+m/z理论值:194.0634,实测值:194.0639。
实施例2-1:3-(2-甲基苄基)噻唑烷-2-酮的合成方法(1)
在反应管中,依次加入邻甲基苄溴(134μl,1mmol)、2-(甲硫基)-4,5-二氢噻唑(56μl,0.5mmol)、叔丁醇钾(56mg,0.05mmol)、碘(256mg,1.0mmol)和碳酸二甲酯(1ml),在反应温度为80℃情况下电磁搅拌反应,反应时间为24小时。反应完成后,将溶剂用旋转蒸发去除,混合物用柱层析分离,洗脱剂为乙酸乙酯和石油醚(体积比为1:9),分离后得到淡黄色液体(89mg,86%)。
实施例2-2:3-(2-甲基苄基)噻唑烷-2-酮的合成方法(2)
在反应管中,依次加入邻甲基苄溴(134μl,1mmol)、2-(甲硫基)-4,5-二氢噻唑(56μl,0.5mmol)、叔丁醇钾(56mg,0.5mmol)、碘(256mg,1.0mmol)和碳酸二甲酯(1ml),在反应温度为50℃情况下电磁搅拌反应,反应时间为24小时。反应完成后,将溶剂用旋转蒸发去除,混合物用柱层析分离,洗脱剂为乙酸乙酯和石油醚(体积比为1:9),分离后得到淡黄色液体(70mg,68%)。
实施例2-3:3-(2-甲基苄基)噻唑烷-2-酮的合成方法(3)
在反应管中,依次加入邻甲基苄溴(134μl,1mmol)、2-(甲硫基)-4,5-二氢噻唑(56μl,0.5mmol)、叔丁醇钾(56mg,0.5mmol)、碘(256mg,1.0mmol)和碳酸二甲酯(1ml),在反应温度为100℃情况下电磁搅拌反应,反应时间为24小时。反应完成后,将溶剂用旋转蒸发去除,混合物用柱层析分离,洗脱剂为乙酸乙酯和石油醚(体积比为1:9),分离后得到淡黄色液体(86mg,85%)。
产物检测数据如下:
1hnmr(600mhz,cdcl3):δ=7.22-7.17(m,4h),4.49(s,2h),3.44(t,2h,j=7.2hz),3.21(t,2h,j=7.2hz),2.31(s,3h)。
13c{1h}nmr(150mhz,cdcl3):δ=171.9,136.9,133.9,130.8,129.0,128.1,126.3,48.0,46.9,25.6,19.2.hrms(ei):c11h14ons[m+h]+m/z理论值:208.0791,实测值:208.0792。
实施例3:3-(4-甲基苄基)噻唑烷-2-酮的合成方法
在反应管中,依次加入对甲基苄溴(185mg,1mmol)、2-(甲硫基)-4,5-二氢噻唑(56μl,0.5mmol)、叔丁醇钾(56mg,0.5mmol)、碘(256mg,1.0mmol)和甲苯(1ml),在反应温度为80℃情况下电磁搅拌反应,反应时间为16小时。反应完成后,将溶剂用旋转蒸发去除,混合物用柱层析分离,洗脱剂为乙酸乙酯和石油醚(体积比为1:9),分离后得到淡黄色液体(169mg,82%)。
产物检测数据如下:
1hnmr(400mhz,cdcl3):δ=7.28-7.17(m,4h),4.45(s,2h),3.51(t,2h,j=7.2hz),3.22(t,2h,j=7.6hz),2.36(s,3h)。
13c{1h}nmr(100mhz,cdcl3):δ=172.2,137.7,133.0,129.5,128.3,128.2,48.5,48.0,25.6,21.2.hrms(ei):c11h14ons[m+h]+m/z理论值:208.0791,实测值:208.0796。
实施例4:3-(4-(叔丁基)苄基)噻唑烷-2-酮的合成方法
在反应管中,依次加入对叔丁基苄溴(185μl,1mmol)、2-(甲硫基)-4,5-二氢噻唑(56μl,0.5mmol)、叔丁醇钾(56mg,0.5mmol)、碘(256mg,1.0mmol)和四氢呋喃(1ml),在反应温度为80℃情况下电磁搅拌反应,反应时间为20小时。反应完成后,将溶剂用旋转蒸发去除,混合物用柱层析分离,洗脱剂为乙酸乙酯和石油醚(体积比为1:9),分离后得到无色固体(112mg,90%)。
产物检测数据如下:
1hnmr(400mhz,cdcl3):δ=7.37(d,2h,j=8.0hz),7.21(d,2h,j=8.4hz),4.47(s,2h),3.53(t,2h,j=7.2hz),3.23(t,2h,j=7.6hz),1.33(s,9h)。
13c{1h}nmr(100mhz,cdcl3):δ=172.2,150.9,133.0,128.0,125.8,48.4,48.1,34.6,31.4,25.5.hrms(ei):c14h20ons[m+h]+m/z理论值:250.1260,实测值:250.1262。
实施例5:3-(4-溴苄基)噻唑烷-2-酮的合成方法
在反应管中,依次加入对溴苄溴(136μl,1mmol)、2-(甲硫基)-4,5-二氢噻唑(56μl,0.5mmol)、叔丁醇钾(56mg,0.5mmol)、碘(256mg,1.0mmol)和乙醇(1ml),在反应温度为80℃情况下电磁搅拌反应,反应时间为20小时。反应完成后,将溶剂用旋转蒸发去除,混合物用柱层析分离,洗脱剂为乙酸乙酯和石油醚(体积比为1:9),分离后得到无色液体(96mg,71%)
产物检测数据如下:
1hnmr(400mhz,cdcl3):δ=7.57(d,2h,j=8.4hz),7.15(d,2h,j=8.4hz),4.43(s,2h),3.50(t,2h,j=7.2hz),3.24(t,2h,j=7.2hz)。
13c{1h}nmr(100mhz,cdcl3):δ=172.4,135.2,132.0,129.9,121.9,48.1,48.0,25.6.hrms(ei):c10h11onbrs[m+h]+m/z理论值:271.9739,实测值:271.9742。
- 还没有人留言评论。精彩留言会获得点赞!