一种苯丙素类化合物及其制备方法和用途与流程
本发明属于天然药物化学领域,具体涉及一种苯丙素类化合物及其制备方法和用途。
背景技术:
荔枝草为唇形科鼠尾草属植物荔枝草(salviaplebeiar.brown)的全草,别名雪见草、癞子草、癞蛤蟆草等,它是一种民间常见中药,味苦、辛、性凉,归肺、胃、肾经,具有清热解毒,利尿消肿,凉血止血等功效。荔枝草含有多种化学成分,主要包括黄酮类化合物、苯丙素类化合物、萜类化合物以及挥发油类化合物。现代药理研究表明,荔枝草具有抗菌消炎、抗氧化、抗病毒、保肝、化痰止咳平喘、降脂减肥等作用。荔枝草在我国分布非常广泛,野生资源极为丰富,是一种极具开发价值的药食两用中药。目前对荔枝草活性成分的研究有限,还有待进一步研究和开发。
技术实现要素:
基于荔枝草丰富的化学成分和多种药理活性,本发明采用生物活性指导分离的方法,从荔枝草中分离得到一种新苯丙素类化合物,可进一步拓展荔枝草的应用。具体技术方案为:所述苯丙素类化合物的名称为:(e)-1-丙烯醛基-3-异丙醇基-7-羟基苯基-8-羟甲基-苯并呋喃,分子式为c21h20o5,具有如下结构:
优选的,所述苯丙素类化合物是从荔枝草中提取得到的。
本发明另一目的在于,提供了上述苯丙素类化合物的制备方法,具体技术方案为:
所述苯丙素化合物的制备方法包括如下步骤:
(1)将荔枝草的乙醇提取物浓缩至无醇味,得到稠膏;
(2)将步骤(1)得到的所述稠膏混悬于水中,得到混悬液,依次用石油醚、乙酸乙酯和正丁醇对所述混悬液进行萃取,先后得到石油醚萃取液、乙酸乙酯萃取液和正丁醇萃取液;
(3)将步骤(2)得到的乙酸乙酯萃取液上硅胶柱,依次用15:1、10:1、8:1、6:1的环己烷-乙酸乙酯洗脱系统对所述硅胶柱进行梯度洗脱,得到4个馏分b-1、b-2、b-3和b-4;
(4)将步骤(3)所得馏分b-2上硅胶柱,依次用13:1、10:1、7:1的石油醚-乙酸乙酯洗脱系统对硅胶柱进行梯度洗脱,得到3个馏分b-2-1、b-2-2和b-2-3。
(5)将步骤(4)所得亚馏分b-2-3上sephadexlh-20凝胶柱,用90%、95%、100%甲醇进行洗脱,得到次馏分b-2-3-1、b-2-3-2和b-2-3-3。
(6)将步骤(5)得次馏分b-2-3-3,进行液相分离纯化,得到所述苯丙素类化合物。
优选的,所述乙醇提取物是通过将荔枝草粉碎至30-50目,用体积分数85-100%乙醇浸泡8-10小时,然后加热回流提取3次,每次5-8小时,合并提取液得到的。
优选的,配制步骤(2)中的所述混悬液时,所述稠膏与水的体积比为1:(2-3)。
优选的,所述步骤(6)中液相的条件为:色谱柱:ymc-packods-a柱(250×20mm,5μm),流动相:30%-50%甲醇,波长:210nm,流速:3-5ml/min。
优选的,所述步骤(2)-(6)中的任意一个或几个步骤还包括采用mtt法进行血管增生抑制活性实验以验证所述苯丙素类化合物是否存在的操作。由于本发明的苯丙素类化合物具有抑制血管增生的作用,在从荔枝草中提取本发明的苯丙素类化合物的过程中,可通过测定不同馏分/洗脱液对血管增生的抑制作用,判断其主要存在于哪些馏分/洗脱液中。
本发明的目的还在于,提供了上述苯丙素类化合物的用途,具体技术方案为:如上述任意一项所述的苯丙素类化合物在制备血管增生抑制剂中的应用。
优选的,如上述任意一项所述的苯丙素类化合物在制备抗肿瘤药物中的应用。
优选的,如上述任意一项所述的苯丙素类化合物在制备抗肾癌药物中的应用。
本发明的有益效果是:本发明从荔枝草中分离得到了一种新的苯丙素类化合物,该苯丙素类化合物具有抑制血管增生的作用,可用于制备血管增生抑制剂/抗肿瘤药物/抗肾癌药物。
本发明的苯丙素类化合物采用现代提取方法,综合运用硅胶柱色谱、凝胶柱色谱和制备液相色谱等分离、纯化手段,提取方法简便快捷、易于操作。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其他的附图。
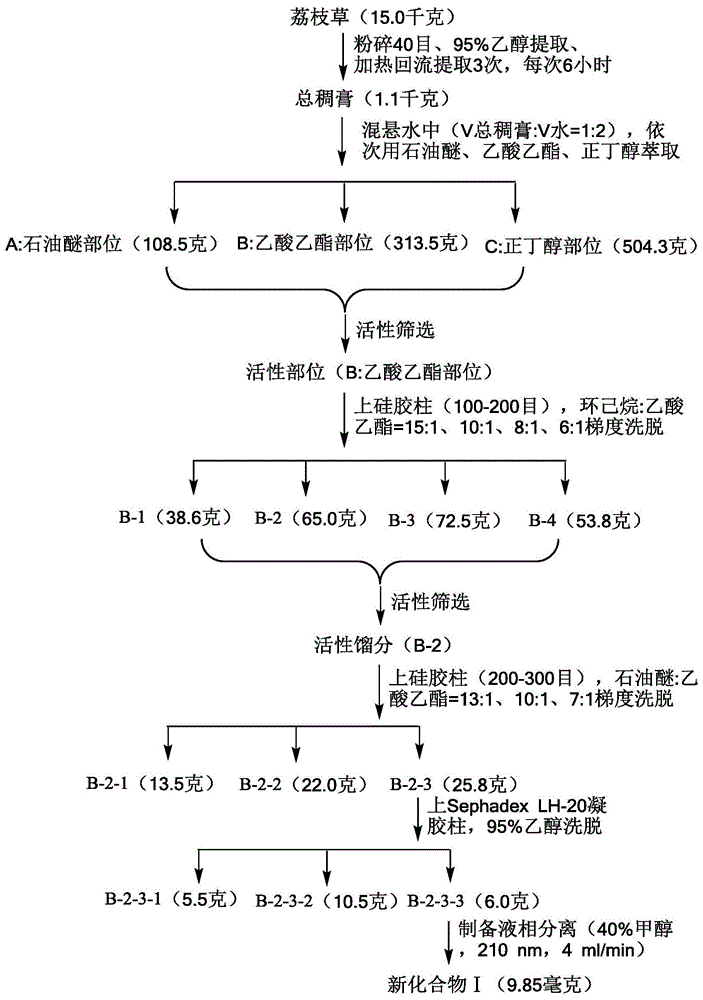
图1为本发明的实施例1的苯丙素类化合物的制备流程图(图1中“新化合物i”代指本发明的苯丙素类化合物、活性筛选的具体步骤同实施例4);
图2为本发明的苯丙素类化合物;
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
下述实施例中所用到的主要试剂及其来源:10%胎牛血清、100u/mg链霉素、100u/ml青霉素、磷酸缓冲盐溶液(pbs)、胰蛋白酶消化液、mtt溶液和dmso溶液等(上海素尔生物科技有限公司);huvec细胞(上海弘顺生物科技有限公司)、rpmi1640培养基(上海博升生物科技有限公司)、阳性对照药:axitinib(上海格物致知医药科技有限公司)。
实施例1
(1)将干燥的荔枝草15.0千克粉碎至40目,在95%乙醇(40l)中浸泡9小时,然后加热回流提取3次,每次6小时,合并提取液,减压浓缩至无醇味,得到总稠膏1.1千克;
(2)将步骤(1)得到的总稠膏混悬于水中(v总稠膏/v水=1:2.5),依次用石油醚(12l)、乙酸乙酯(11l)、正丁醇(10l)进行萃取,分别得到石油醚部位(a:108.5克)、乙酸乙酯部位(b:313.5克)、正丁醇部位(c:504.3克);
(3)根据生物活性指导分离的方法,采用mtt法对上述不同萃取部位进行血管增生抑制活性筛选,确定乙酸乙酯部位(b)为活性部位;
(4)将步骤(3)得到的活性部位(b)上硅胶(100-200目)柱,依次用环己烷-乙酸乙酯(15:1、10:1、8:1、6:1,体积比,根据tlc检测确定溶剂量)洗脱系统进行梯度洗脱,先后得到4个馏分b-1(38.6克)、b-2(65.0克)、b-3(72.5克)、b-4(53.8克);
(5)结合前期活性筛选结果,将活性馏分b-2再次上硅胶(200-300目)柱,依次用石油醚-乙酸乙酯(13:1、10:1、7:1,体积比,根据tlc检测确定溶剂量)洗脱系统进行梯度洗脱先后得到亚馏分b-2-1(13.5克)、b-2-2(22.0克)、b-2-3(25.8克);
(6)将步骤(5)得到的亚馏分b-2-3上sephadexlh-20凝胶柱,依次用90%(体积分数)、95%(体积分数)、100%甲醇进行洗脱(根据tlc检测确定溶剂量),先后得到次馏分b-2-3-1(5.5克)、b-2-3-2(10.5克)、b-2-3-3(6.0克);
(7)将步骤(6)得到的次馏分b-2-3-3,进行制备液相(色谱柱:ymc-packods-a柱(250×20mm,5μm)、流动相:40%甲醇、波长:210nm、流速:4ml/min)分离,得到苯丙素类化合物(9.85毫克)。
本发明所述的苯丙素类化合物为黄色粉末,hr-esi-msm/z374.1149[m+na]+揭示其分子式为c21h20o5(calcd.forc21h19o5na,374.1128)。本发明的苯丙素类化合物的uv(meoh)λmax:210、330nm;irνmax:3378、1766、1633、1520、975cm-1;1hnmr(cd3cocd3,400mhz)和13cnmr(cd3cocd3,100mhz)数据见表1。
根据本发明的苯丙素类化合物的波谱数据和hmbc和1h-1hcosy偶联相关信息(详见图2),经scifinder检索,确定本发明的苯丙素类化合物为一个新苯丙素类化合物。
表1本发明的苯丙素类化合物的1hnmr、13cnmr和hmbc相关数据
实施例2
(1)将干燥的荔枝草粉碎至50目,在90%乙醇(40l)中浸泡8小时,然后加热回流提取3次,每次5小时,合并提取液,减压浓缩至无醇味,得到总稠膏1.0千克;
(2)将步骤(1)得到的总稠膏混悬于水中(v总稠膏/v水=1:2),依次用石油醚(12l)、乙酸乙酯(11l)、正丁醇(10l)进行萃取,分别得到石油醚部位(a:106.0克)、乙酸乙酯部位(b:308.5克)、正丁醇部位(c:500.2克);
(3)根据生物活性指导分离的方法,采用mtt法对上述不同萃取部位进行血管增生抑制活性筛选,确定乙酸乙酯部位(b)为活性部位;
(4)将步骤(3)得到的活性部位(b)上硅胶(100-200目)柱,依次用环己烷-乙酸乙酯(15:1、12:1、9:1、6:1,体积比,根据tlc检测确定溶剂量)洗脱系统进行梯度洗脱,先后得到4个馏分b-1(35.5克)、b-2(60.3克)、b-3(71.0克)、b-4(50.5克);
(5)结合前期活性筛选结果,将活性馏分b-2再次上硅胶(200-300目)柱,依次用石油醚-乙酸乙酯(13:1、9:1、7:1,体积比)洗脱系统进行梯度洗脱先后得到亚馏分b-2-1(12.4克)、b-2-2(20.5克)、b-2-3(24.5克);
(6)将步骤(5)得到的亚馏分b-2-3上sephadexlh-20凝胶柱,依次用90%(体积分数)、96%(体积分数)、100%甲醇进行洗脱(根据tlc检测确定溶剂量),先后得到次馏分b-2-3-1(4.8克)、b-2-3-2(9.5克)、b-2-3-3(5.0克);
(7)将步骤(6)得到的次馏分b-2-3-3,进行制备液相(色谱柱:ymc-packods-a柱(250×20mm,5μm)、流动相:30%甲醇、波长:210nm、流速:3ml/min)分离,得到苯丙素类化合物(9.70毫克)。
经验证,实施例2制备得到了与实施例1结构相同的化合物。
实施例3
(1)将干燥的荔枝草粉碎至30目,在85%乙醇(体积分数)中浸泡10小时,然后加热回流提取3次,每次8小时,合并提取液,减压浓缩至无醇味,得到总稠膏(1.2千克);
(2)将步骤(1)得到的总稠膏混悬于水中(v总稠膏/v水=1:3),依次用石油醚(12l)、乙酸乙酯(11l)、正丁醇(10l)进行萃取,分别得到石油醚部位(a:110.0克)、乙酸乙酯部位(b:315.4克)、正丁醇部位(c:506.0克);
(3)根据生物活性指导分离的方法,采用mtt法对上述不同萃取部位进行血管增生抑制活性筛选,确定乙酸乙酯部位(b)为活性部位;
(4)将步骤(3)得到的活性部位(b)上硅胶(100-200目)柱,依次用环己烷-乙酸乙酯(15:1、11:1、8:1、6:1,体积比,根据tlc检测确定溶剂量)洗脱系统进行梯度洗脱,先后得到4个馏分b-1(39.0克)、b-2(67.5克)、b-3(74.5克)、b-4(54.3克);
(5)结合前期活性筛选结果,将活性馏分b-2再次上硅胶(200-300目)柱,依次用石油醚-乙酸乙酯(13:1、11:1、7:1,体积比,根据tlc检测确定溶剂量))洗脱系统进行梯度洗脱先后得到亚馏分b-2-1(14.9克)、b-2-2(23.5克)、b-2-3(27.0克);
(6)将步骤(5)得到的亚馏分b-2-3上sephadexlh-20凝胶柱,依次用90%(体积分数)、94%(体积分数)、100%甲醇进行洗脱(根据tlc检测确定溶剂量),先后得到次馏分b-2-3-1(6.0克)、b-2-3-2(11.6克)、b-2-3-3(7.2克);
(7)将步骤(6)得到的次馏分b-2-3-3,进行制备液相分离,(流动相:50%甲醇、波长:210nm、流速:5ml/min,得到苯丙素类化合物(9.96毫克)。
经验证,实施例3制备得到了与实施例1结构相同的苯丙素类化合物。
实施例4血管增生抑制活性筛选
4.1细胞培养与传代
将对数生长期的huvec细胞分别接种于含有10%胎牛血清、100u/mg链霉素、100u/ml青霉素的rpmi1640培养皿中,在37℃、5%co2培养箱中进行培养。当培养皿细胞贴壁达80%左右时,吸出原培养液,换用磷酸缓冲盐溶液(pbs)清洗细胞2次,然后吸出pbs,加入1ml胰蛋白酶消化液,在显微镜下观察细胞消化状态,若细胞回缩变圆、透亮、轻拍培养皿呈流沙样脱落,或大部分细胞已成单细胞悬液,则加入含10%胎牛血清的完全培养液终止消化,并轻轻吹打细胞,使其完全成单细胞悬液后,2000r/min离心5min,除去上清液,加入培养液混悬,按1:3比例接种至新培养皿中,置于培养箱中继续培养,每日观察细胞生长状态,约2-3天换液1次,当细胞贴壁达85%-95%时传代,传至第3代后细胞状态稳定时,用于后续实验。
4.2mtt法测定细胞增殖
4.2.1取对数生长期的huvec细胞,胰酶消化离心后收集细胞,采用rpmi1640完全培养液重悬,计数并调整细胞密度为2.5×104个/ml,取200μl重悬液接种于96孔板中,置于37℃、5%co2培养箱中24小时。
4.2.2除去上清液,加入含有本发明的苯丙素类化合物或axitinib的培养液,200μl/孔,使其终浓度分别为1.0、2.0、4.0、8.0、16.0、32.0mg/ml,继续培养72小时。
4.2.3培养结束后,毎孔加5mg/mlmtt溶液20μl,在37℃继续培养4小时,弃上清液,每孔加入dmso溶液150μl,振荡10min,充分溶解结晶,全自动酶标仪测定各孔490nm处a值。
4.2.4计算增殖抑制率:抑制率(%)=(1-实验组a490/对照组a490)×100%和ic50值(注:axitinib为阳性对照药)。
4.3实验结果
本发明通过上述方法对本发明的苯丙素类化合物进行血管增生抑制活性筛选,试验结果表明本发明的苯丙素类化合物具有显著的血管增生抑制活性(表2)。
表2本发明的苯丙素类化合物/axitinib对huvec细胞增殖抑制活性筛选结果(n=5)
其中,实施例1、2、3及axitinib按照上述4.2部分的方法分别实验,上表2中的“本发明的苯丙素类化合物”为实施例1、2、3(每个实施例做5个重复)各组实验测得的ic50平均值;上表2中的axitinib表示5组实验测得的ic50平均值。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!