核苷酸序列构建的CRISPR重组质粒靶向编辑ISS序列的方法与流程
本发明属于生物技术领域,具体设计一种sgrnaqingqinoligo核苷酸序列构建的crispr重组质粒靶向编辑iss序列的方法。
背景技术:
smn2基因外显子7列入对于脊髓性肌肉萎缩症(sma)病人的重要性:研究表明sma是由于第五号染色体上运动神经元生存1(survivalofmotorneuron1,smn1)基因发生点突变或缺失导致该基因不能表达有功能的全长smn蛋白。和动物不同,人类因为染色体5q13区的倒转复制(invertedduplication)产生了一个smn1的平行同源基因称作smn2;而两个基因包含9个外显子(1、2a、2b、3、4、5、6、7、8),编码完全相同294个氨基酸的smn蛋白。两者的关键不同之处在于外显子7第6位核苷酸由smn1中的c转变为smn2中的t(c6t),虽然没有造成翻译中氨基酸的改变,却严重影响了外显子7的列入(exon7inclusion)。smn2的成熟转录产物中约有90%不包含外显子7,而没有外显子7的蛋白(称作smnδ7)基本没有功能且极不稳定。smn2表达的少量全长smn蛋白虽然不足以补偿smn1基因的缺陷,却对病人的生存至关重要。
crispr/cas9精准靶向编辑基因改善基因功能的相关研究:随着生物技术的飞速发展,crispr/cas9基因编辑技术及基因治疗已成为研究热点。近年来国际权威期刊《科学》杂志(science),频繁报道crispr/cas9基因编辑技术应用于型肌营养不良(duchennemusculardystrophy,dmd)基因治疗的论文,以dmd动物模型-mdx小鼠为研究对象,进行动物体内试验。借助aav载体运载基因编辑的crispr/cas9进入体内细胞,做删除式基因编辑。删除了含有无义突变的23号外显子,形成永久性不含外显子23的dmd基因。治疗后肌肉病理得到改善,肌肉细胞膜上重新出现dystrophin蛋白,小鼠肌肉力量得到增加。但是该项技术在sma小鼠的治疗中尚没有相关研究报道,急需要开展新方法的探索研究。
在人类体内表达smn蛋白有两个基因,smn1和smn2,但是起主要作用的smn1基因突变失去功能后,由于smn2基因外显子7大部分被剪切而仅有少量smn全长基因,少量smn有功能蛋白,目前最有效的方法是aso反义寡核苷酸,精准靶向互补内含子7第10-27序列,比如16年在美国上市的寡核苷酸药物spinraza,靶向封闭内含子7剪接沉默子iss序列,从而显著促进外显子7列入比例,但是aso半衰期较短,必须持续性给药。
技术实现要素:
本发明要解决的技术问题是提供一种核苷酸序列构建的crispr重组质粒靶向编辑iss序列的方法,采用crisprcase9sgrnaqingqin重组质粒,可以精准靶向编辑(包括删除、插入及突变)iss序列,也可以达到使剪接沉默子iss序列失去作用或作用减弱的效果,从而显著促进smn2外显子7列入,而且该重组质粒可以整合入宿主细胞基因组内,在细胞内持续性表达,一次性转染即可。
为解决上述技术问题,本发明的实施例提供一种核苷酸序列构建的crispr重组质粒靶向编辑iss序列的方法,步骤为:靶向设计sgrnaqingqinoligo序列,两端加商业化质粒酶切位点,构建重组质粒,转化,铺板,挑单克隆,提质粒,测序,序列比对,经鉴定,构建成功。
上述的核苷酸序列构建的crispr重组质粒靶向编辑iss序列的方法包括如下具体步骤:
(1)bsmbi酶切lenticrispr空载:restrietionenzyme1ul,质粒dna5ug,10×buffer5ul,补灭菌去离子水至50ul,55℃酶切30min,然后80℃,酶切20min;
(2)将步骤(1)的酶切产物纯化备用;
(3)目标oligosgrna退火:sgrnaf(100um)2ul,sgrnar(100um)2ul,10×t4ligationbuffer2ul,t4pnk1ul,补灭菌去离子水至20ul,37℃退火30min,然后95℃退火5min,每分钟降1~6℃,梯度退至20℃火;
(4)将步骤(3)退火后的oligosgrna在1:50~1:100之间稀释;
(5)链接反应:步骤(1)的bsmbi酶切lenticrispr2~8ul,步骤(4)的oligosgrna1~3ul,10×t4ligationbuffer1~3ul,t4ligase1~3ul,补灭菌去离子水至20ul,4℃过夜链接;
(6)取步骤(5)的链接产物2~8ul,与100ul感受态细胞混匀,冰浴10min,42℃热激90s,冰浴30min,加入lb液体培养基200ul,200rpm搅拌45min,取120ul涂氨苄板,37℃倒置培养过夜;
(7)挑取单个菌斑与枪头一起放入15ml离心管,加入3~8ml(含1/1000氨苄)lb液培,37℃,200rpm过夜摇菌;
(8)质粒提取:按照axyprepplasmidminiprepkit说明书操作提取质粒,od值检测质粒浓度,然后送公司测序;
(9)对测序结构进行分析,鉴定sgrnaqingqinoligo核苷酸序列构建的crispr重组质粒。
其中,构建的crispr重组质粒选用:
sgrnaqingqinoligo核苷酸序列(5’caccgaagattcactttcataatgc3’,3’cttctaagtgaaagtattacgcaaa5’)。
步骤(2)中利用axygenaxypreppcrcleanupkitforgenonomicdnapurification试剂盒将酶切产物纯化。
本发明的上述技术方案的有益效果如下:本发明采用crisprcase9sgrnaqingqin重组质粒,可以精准靶向编辑(包括删除、插入及突变)iss序列,也可以达到使剪接沉默子iss序列失去作用或作用减弱的效果,从而显著促进smn2外显子7列入,而且该重组质粒可以在细胞内持续性表达,一次性转染即可。
附图说明
图1为本发明的工艺流程图;
图2为本发明中进行pcr测序时的示意图;
图3为本发明中sgrnaqingqin精准靶向编辑iss序列从而显著促进smn2外显子7列入比例的示意图;
图4为在脊髓性肌肉萎缩小鼠原代培养软骨细胞中sgrnaqingqin重组质粒促进smn2外显子7列入比例的示意图。
具体实施方式
为使本发明要解决的技术问题、技术方案和优点更加清楚,下面将结合附图及具体实施例进行详细描述。
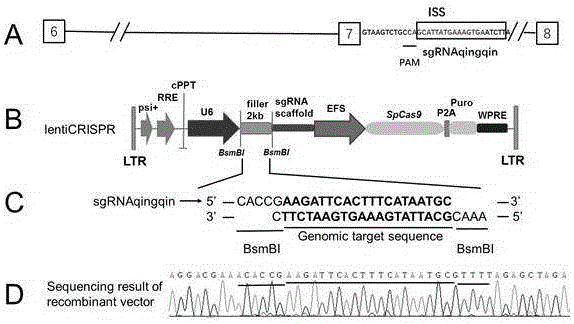
本发明提供一种sgrnaqingqinoligo核苷酸序列构建的crispr重组质粒精准靶向编辑iss序列的方法,sgrnaqingqinoligo构建的crispr重组质粒能够精准靶向smn2内含子7剪接沉默子iss序列(图1a红色斜体),该沉默子附近恰好存在crispr能够识别的ngg对应序列(图1a下划线序列),而且通过基因组序列查重,设计的sgrnaqingqin序列(图1a黑色方框内)在人类基因组内是唯一的,也就是特异性序列。本发明的步骤为:靶向设计sgrnaqingqinoligo序列(图1c),两端加商业化质粒酶切位点(图1bbsmbi),构建重组质粒,转化,铺板,挑单克隆,提质粒,测序,序列比对,经鉴定,构建成功(图1d)。
本发明的sgrnaqingqinoligo核苷酸序列构建的crispr重组质粒精准靶向编辑iss序列的方法包括如下具体步骤:
(1)bsmbi酶切lenticrispr空载:restrietionenzyme1ul,质粒dna5ug,10×buffer5ul,补灭菌去离子水至50ul,55℃酶切30min,然后80℃,酶切20min;
(2)利用axygenaxypreppcrcleanupkitforgenonomicdnapurification试剂盒将步骤(1)的酶切产物纯化备用;
(3)目标oligosgrna退火:sgrnaf(100um)2ul,sgrnar(100um)2ul,10×t4ligationbuffer2ul,t4pnk1ul,补灭菌去离子水至20ul,37℃退火30min,95℃退火5min,每分钟降3℃,梯度退火至20℃;
(4)将步骤(3)退火后的oligosgrna在1:50~1:100之间稀释;
(5)链接反应:步骤(1)的bsmbi酶切lenticrispr4ul,步骤(4)的oligosgrna1~3ul,10×t4ligationbuffer2ul,t4ligase2ul,补灭菌去离子水至20ul,4℃过夜链接;
(6)取步骤(5)的链接产物4ul,与100ultop10感受态混匀,冰浴10min,42℃热激90s(将质粒转入感受态细胞),冰浴30min,加入lb液体培养基200ul,200rpm搅拌45min,取120ul涂氨苄板,37℃倒置培养过夜;
(7)挑取单个菌斑与枪头一起放入15ml离心管,加入约5ml(含1/1000氨苄)lb液培,37℃,200rpm过夜摇菌;
(8)质粒提取:按照axyprepplasmidminiprepkit说明书操作提取质粒,od值检测质粒浓度,然后送公司测序;
(9)对测序结构进行分析,鉴定sgrnaqingqinoligo核苷酸序列构建的crispr重组质粒靶向编辑iss序列,即鉴定目标sgrna序列是否插入载体。
下面通过具体实施例进一步阐述本发明的技术方案。
转染hek293细胞,用puromycin筛选稳定株,提dna进行pcr(图2a)测序,证明sgrna2系统发挥了case9对smn2基因组有编辑的作用,箭头向后有碱基的缺失或者插入(图2b),从pam位点向下游有套峰的出现,通过ta克隆蓝白斑筛选,单克隆测序,结果表明基因编辑的部位恰好是我们设计的sgrnangg下游smn2剪接沉默子(iss)区域(图2c和图2d),以上证明本发明构建的重组质粒精准靶向编辑成功。而且通过pcr电泳,加入3ulddei酶切过夜,2%琼脂糖凝胶电泳检测,灰度值统计学分析,按照以下公式:
统计学分析结果显示exon7列入比例提高约10%,差异具有统计学意义(图3a和图3b),smn2基因外显子7列入比例提高了近10个百分点,而且,在脊髓性肌肉萎缩小鼠原代培养软骨细胞中sgrnaqingqin重组质粒从而显著促进smn2全长基因的表达(图4a和图4b)。
本发明sgrnaqingqin构建的crispr重组质粒可以精准靶向编辑iss序列(删除、插入及突变),可以达到使剪接沉默子iss序列失去作用或作用减弱的效果,从而显著促进smn2外显子7的列入,该方法从基因组水平编辑,从决定蛋白表达最根本的基因组水平纠正,并且重组质粒可以整合到基因组中持续表达。
本发明关键点是设计的sgrnaqingqinoligo核苷酸序列构建的crispr重组质粒能够精准靶向编辑iss序列,从而显著促进smn2基因外显子7的例如比例。对于通过改变sgrnaqingqin序列中的某个或者少量碱基也能起到相同作用,所以碱基和该序列排序以及重复率也应视为本发明的保护范围。
以上所述是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明所述原理的前提下,还可以作出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!