1,3,4-噁二唑-2-环丁基类化合物及其制备方法和应用与流程
本发明属于医药化学领域,具体涉及具有1,3,4-噁二唑-2-环丁基类结构的化合物或其药学可接受的盐、水合物、溶剂合物或结晶及其制备方法和它们在制备治疗癌症或者感染类疾病的药物中的应用。
背景技术:
程序性死亡受体-1(programmedcelldeath-1,pd-1)及其配体pd-ll(b7·h1)属于cd28/b7超家族。pd-1主要表达在t细胞、b细胞、自然杀伤细胞(natualkillercell,nk细胞)的膜表面,pd-l1主要表达于成熟的cd4t细胞、cd8t细胞、b细胞、单核细胞、树突状细胞(dendriticcells,dcs)、巨噬细胞等造血细胞及一些非造血细胞,如内皮细胞、胰岛细胞、肥大细胞等的膜表面。其中pd-l1在多种肿瘤中高表达,如肺癌、胃癌、多发性骨髓、黑色素瘤和乳腺癌等。肿瘤细胞表面上的pd-l1的表达与t细胞表面的配体相互作用,可诱导t细胞的凋亡或降低t细胞的反应活性,从而抑制肿瘤免疫应答,使肿瘤细胞逃避免疫攻击。因此阻断pd1-pdl1信号通路的拮抗剂,可促进t细胞的激活、逆转肿瘤免疫微环境、增强内源性抗肿瘤免疫效应。靶向pd-1/pd-l1抑制剂在肿瘤免疫治疗领域有着广阔的应用前景。
目前抗pd-1/pd-l1抗体治疗在临床上已显示具有优势作用,然而生物大分子也具有一些缺点,例如免疫原性以及给药途径的限制。因此,仍需要开发具有更好药效的靶向pd-1/pd-l1抑制剂。本发明的发明人发现一种小分子药物能特异性地调控和/或调解pd-l1及其相关蛋白激酶的转导,从而用于治疗与pd-1/pd-l1相关的疾病。
技术实现要素:
本发明的一个目的是提供式(i)所示的(((1r,3s)-1-(5-((1s,2r)-1-氨基-2-羟丙基)-1,3,4-噁二唑-2-基)-3-(氰甲基)环丁基)氨基甲酰基)-l-丝氨酸或其药学可接受的盐、水合物或溶剂合物,
在一些具体的实施方案中,本发明提供式(i)的化合物或其药学可接受的盐、水合物或溶剂合物的制备方法,其中所述方法包括如下步骤:
1)式1的化合物和式2的化合物发生缩合反应制得式3的化合物;
2)式3的化合物在脱水剂的作用下发生闭环反应制得式4的化合物;
3)式4的化合物脱除氨基保护基制得式5的化合物;
4)式5的化合物和式6的化合物发生反应制得式7的化合物;
5)式7的化合物发生反应制得式8的化合物;
6)式8的化合物与碱性试剂反应制得式9的化合物;
7)式9的化合物脱除氨基保护基制得式(i)的化合物。
在一些具体的实施方案中,本发明提供式1的化合物的制备方法,其中所述方法包括如下步骤:
1)式1a的化合物的羧基发生酯化反应制得式1b的化合物;
2)式1b的化合物的氨基发生反应制得式1c的化合物;
3)式1c的化合物的羟基发生反应制得式1d的化合物;
4)式1d的化合物发生反应制得式1e的化合物;
5)式1e的化合物发生反应制得式1的化合物。
在一些具体的实施方案中,本发明提供式2的化合物的制备方法,其中所述方法包括如下步骤:
1)式2a的化合物和式2b的化合物发生反应制得式2c的化合物;
2)式2c的化合物和式2d的化合物发生反应制得式2e的化合物;
3)式2e的化合物脱除氨基保护基制得式2的化合物。
在一些具体的实施方案中,根据本发明的式(i)的化合物或其药学可接受的盐、水合物或溶剂合物的制备方法,其中所述脱水剂选自三甲基氯硅烷、三氟甲磺酰氯、多聚磷酸、4-甲基苯磺酰氯、碘/三苯基膦、三氯氧磷、亚硫酰氯、磷酰氯、hcl、二氯三苯基正膦、hoac、硫酸和乙酸酐。
本发明的另一个目的是提供式(i)所示的(((1r,3s)-1-(5-((1s,2r)-1-氨基-2-羟丙基)-1,3,4-噁二唑-2-基)-3-(氰甲基)环丁基)氨基甲酰基)-l-丝氨酸的晶型。
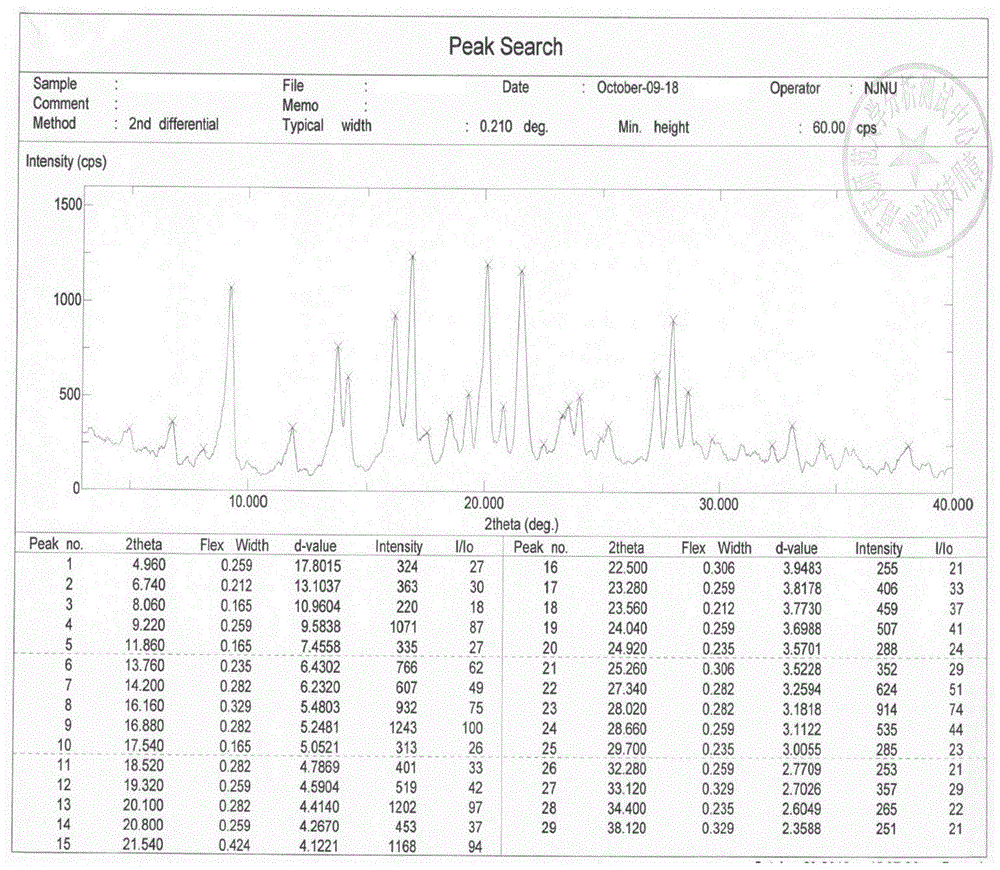
本发明一方面提供式(i)化合物晶型a。在一些优选的实施方案中,本发明式(i)化合物的晶型a的x-射线粉末衍射图谱,使用cu-ka辐射,以2θ角度表示x-射线粉末衍射图,其在约6.740±0.2、9.220±0.2、11.860±0.2、16.880±0.2、20.100±0.2处有特征峰;
优选地,本发明式(i)化合物的晶型a的x-射线粉末衍射图谱在约6.740±0.2、9.220±0.2、11.860±0.2、13.760±0.2、16.880±0.2、20.100±0.2、21.540±0.2处有特征峰;
进一步优选地,本发明式(i)化合物的晶型a的x-射线粉末衍射图谱在约4.960±0.2、6.740±0.2、9.220±0.2、11.860±0.2、13.760±0.2、16.880±0.2、20.100±0.2、21.540±0.2处有特征峰;
再进一步优选地,本发明式(i)化合物的晶型a的x-射线粉末衍射图谱在约4.960±0.2、6.740±0.2、8.060±0.2、9.220±0.2、11.860±0.2、13.760±0.2、16.880±0.2、20.100±0.2、21.540±0.2、28.020±0.2处有特征峰;
再进一步优选地,本发明式(i)化合物的晶型a的x-射线粉末衍射图谱在约4.960±0.2、6.740±0.2、8.060±0.2、9.220±0.2、11.860±0.2、13.760±0.2、14.200±0.2、16.160±0.2、16.880±0.2、20.100±0.2、21.540±0.2、27.340±0.2、28.020±0.2处有特征峰;
更进一步优选地,本发明式(i)化合物的晶型a的x-射线粉末衍射图谱在约4.960±0.2、6.740±0.2、8.060±0.2、9.220±0.2、11.860±0.2、13.760±0.2、14.200±0.2、16.160±0.2、16.880±0.2、17.540±0.2、18.520±0.2、19.320±0.2、20.100±0.2、20.800±0.2、21.540±0.2、22.500±0.2、23.280±0.2、23.560±0.2、24.040±0.2、24.920±0.2、25.260±0.2、27.340±0.2、28.020±0.2、28.660±0.2、29.700±0.2、32.280±0.2、33.120±0.2、34.400±0.2、38.120±0.2处有特征峰。
非限制性地,在一个具体的实施方案中,本发明式(i)化合物的晶型a具有如图1所示的x-射线粉末衍射图谱。
本发明提供一种所述的式(i)所示的(((1r,3s)-1-(5-((1s,2r)-1-氨基-2-羟丙基)-1,3,4-噁二唑-2-基)-3-(氰甲基)环丁基)氨基甲酰基)-l-丝氨酸晶型a的制备方法,包括将(((1r,3s)-1-(5-((1s,2r)-1-氨基-2-羟丙基)-1,3,4-噁二唑-2-基)-3-(氰甲基)环丁基)氨基甲酰基)-l-丝氨酸溶于有机溶剂中,析出结晶的步骤。
在一些优选的实施方案中,本发明提供一种所述的式(i)所示的(((1r,3s)-1-(5-((1s,2r)-1-氨基-2-羟丙基)-1,3,4-噁二唑-2-基)-3-(氰甲基)环丁基)氨基甲酰基)-l-丝氨酸晶型a的制备方法,具体包括如下步骤:
(1)将(((1r,3s)-1-(5-((1s,2r)-1-氨基-2-羟丙基)-1,3,4-噁二唑-2-基)-3-(氰甲基)环丁基)氨基甲酰基)-l-丝氨酸溶于有机溶剂中反应,析出结晶;和
(2)过滤、洗涤、干燥。
上述反应步骤(1)中对原料(((1r,3s)-1-(5-((1s,2r)-1-氨基-2-羟丙基)-1,3,4-噁二唑-2-基)-3-(氰甲基)环丁基)氨基甲酰基)-l-丝氨酸的存在形态没有特别限定,可以使用任意晶体或无定型固体。
上述反应步骤(1)中的有机溶剂为有机溶剂或它们的混合溶液,所述有机溶剂选自碳原子数小于6的醇类、酮类、酯类、醚类、腈类、烃类溶剂和四氢呋喃中的一种或多种,所述有机溶剂优选选自甲醇、乙醇、正丙醇、异丙醇、正丁醇、正戊醇、正己醇、丙酮、甲基乙基酮、甲基异丁基酮、乙酸乙酯、乙醚、乙腈、环己烷和四氢呋喃中的一种或多种,更优选选自甲醇、乙醇、异丙醇、正丁醇、乙醚、环己烷、丙酮和四氢呋喃中的一种或多种。
按照本发明的方法制备得到的(((1r,3s)-1-(5-((1s,2r)-1-氨基-2-羟丙基)-1,3,4-噁二唑-2-基)-3-(氰甲基)环丁基)氨基甲酰基)-l-丝氨酸晶型a不含有或者含有较低含量的残留溶剂。
本发明另一方面提供式(i)化合物的晶型b。
在一些实施方案中,本发明式(i)化合物的晶型b的x-射线粉末衍射图谱,使用cu-ka辐射,以2θ角度表示x-射线粉末衍射图,其在约4.900±0.2、9.220±0.2、9.980±0.2、17.380±0.2、20.040±0.2处有特征峰;
优选地,本发明式(i)化合物的晶型b的x-射线粉末衍射图谱在约4.900±0.2、6.680±0.2、9.220±0.2、9.980±0.2、14.220±0.2、16.840±0.2、17.380±0.2、20.040±0.2、21.540±0.2处有特征峰;
进一步优选地,本发明式(i)化合物的晶型b的x-射线粉末衍射图谱在约4.900±0.2、6.680±0.2、8.100±0.2、9.220±0.2、9.980±0.2、14.220±0.2、16.840±0.2、17.380±0.2、20.040±0.2、21.540±0.2、28.000±0.2处有特征峰;
再一步优选地,本发明式(i)化合物的晶型b的x-射线粉末衍射图谱在约4.900±0.2、6.680±0.2、8.100±0.2、9.220±0.2、9.980±0.2、11.820±0.2、14.220±0.2、15.280±0.2、16.220±0.2、16.840±0.2、17.380±0.2、20.040±0.2、21.540±0.2、27.340±0.2、28.000±0.2处有特征峰;
更进一步优选地,本发明式(i)化合物的晶型b的x-射线粉末衍射图谱在约4.900±0.2、6.680±0.2、8.100±0.2、9.220±0.2、9.980±0.2、11.820±0.2、13.360±0.2、13.720±0.2、14.220±0.2、15.280±0.2、16.220±0.2、16.840±0.2、17.380±0.2、18.460±0.2、19.260±0.2、20.040±0.2、21.540±0.2、22.460±0.2、23.200±0.2、23.980±0.2、25.280±0.2、26.120±0.2、27.340±0.2、28.000±0.2、28.520±0.2、29.680±0.2、30.280±0.2、32.180±0.2、33.100±0.2、35.920±0.2处有特征峰。
非限制性地,在一个具体的实施方案中,本发明式(i)化合物的晶型b具有如图2所示的x-射线粉末衍射图谱。
本发明提供另一种所述的式(i)所示的(((1r,3s)-1-(5-((1s,2r)-1-氨基-2-羟丙基)-1,3,4-噁二唑-2-基)-3-(氰甲基)环丁基)氨基甲酰基)-l-丝氨酸晶型b的制备方法,为单一溶剂晶浆结晶法。在一些具体的实施方案中,所述方法包括例如将(((1r,3s)-1-(5-((1s,2r)-1-氨基-2-羟丙基)-1,3,4-噁二唑-2-基)-3-(氰甲基)环丁基)氨基甲酰基)-l-丝氨酸溶于有机溶剂中得到悬浊液的步骤。在一些优选的实施方案中,所述方法进一步包括搅拌的步骤。优选地,所述有机溶剂选自甲醇、乙醇、异丙醇、正丁醇、丙酮、异丙醚、乙酸乙酯、乙酸异丙酯、水饱和的乙酸乙酯、四氢呋喃、1,4-二氧六环、乙腈、氯仿、甲苯和正庚烷中的一种或多种。
按照本发明的方法制备得到的(((1r,3s)-1-(5-((1s,2r)-1-氨基-2-羟丙基)-1,3,4-噁二唑-2-基)-3-(氰甲基)环丁基)氨基甲酰基)-l-丝氨酸晶型b不含有或者含有较低含量的残留溶剂。
本发明的另一方面提供一种药物组合物,其含有(((1r,3s)-1-(5-((1s,2r)-1-氨基-2-羟丙基)-1,3,4-噁二唑-2-基)-3-(氰甲基)环丁基)氨基甲酰基)-l-丝氨酸或其药学可接受的盐、水合物、溶剂合物或结晶及药学上可接受的载体。
本发明的另一方面提供(((1r,3s)-1-(5-((1s,2r)-1-氨基-2-羟丙基)-1,3,4-噁二唑-2-基)-3-(氰甲基)环丁基)氨基甲酰基)-l-丝氨酸或其药学可接受的盐、水合物、溶剂合物或结晶或包含其的药物组合物在制备用于治疗癌症或感染性疾病的药物中的应用。
在一个优选的实施方案中,本发明提供(((1r,3s)-1-(5-((1s,2r)-1-氨基-2-羟丙基)-1,3,4-噁二唑-2-基)-3-(氰甲基)环丁基)氨基甲酰基)-l-丝氨酸或其药学可接受的盐、水合物、溶剂合物或结晶或包含其的药物组合物在制备用于治疗癌症或感染性疾病的药物中的应用,其中所述癌症包括但不限于黑色素瘤、脑瘤(具有恶性的星形神经胶质和少突神经胶质细胞瘤成分的神经胶质瘤等)、食管癌、胃癌、肝癌、胰腺癌、结肠直肠癌(结肠癌、直肠癌等)、肺癌(非小细胞肺癌、小细胞肺癌、原发或转移性鳞状癌等)、肾癌、乳腺癌、卵巢癌、前列腺癌、皮肤癌、神经母细胞瘤、肉瘤、骨软骨瘤、骨瘤、骨肉瘤、精原细胞瘤、睾丸肿瘤、子宫癌(子宫颈癌、子宫内膜癌等)、头颈肿瘤(上颌骨癌、喉癌、咽癌、舌癌、口内癌等)、多发性骨髓瘤、恶性淋巴瘤(网状细胞肉瘤、淋巴肉瘤、霍奇金淋巴瘤等)、真性红细胞增多症、白血病(急性粒细胞白血病、慢性粒细胞白血病、急性淋巴细胞白血病、慢性淋巴细胞白血病等)、甲状腺肿瘤、输尿管肿瘤、膀胱肿瘤、胆囊癌、胆管癌、绒毛膜上皮癌或儿科肿瘤(尤因家族性肉瘤、维尔姆斯肉瘤、横纹肌肉瘤、血管肉瘤、胚胎睾丸癌、成神经细胞瘤、视网膜母细胞瘤、肝胚细胞瘤、肾母细胞瘤等)和所述癌症的组合。
在另一个优选的实施方案中,本发明提供(((1r,3s)-1-(5-((1s,2r)-1-氨基-2-羟丙基)-1,3,4-噁二唑-2-基)-3-(氰甲基)环丁基)氨基甲酰基)-l-丝氨酸或其药学可接受的盐、水合物、溶剂合物或结晶或包含其的药物组合物在制备用于治疗癌症或感染性疾病的药物中的应用,其中所述感染性疾病包括但不限于细菌、病毒和真菌感染。
在一个具体的实施方案中,本发明提供的(((1r,3s)-1-(5-((1s,2r)-1-氨基-2-羟丙基)-1,3,4-噁二唑-2-基)-3-(氰甲基)环丁基)氨基甲酰基)-l-丝氨酸或其药学可接受的盐、水合物、溶剂合物或结晶对结肠癌具有显著的抑制作用。
在这里需要特别说明的是,x-射线粉末衍射图谱对于特定的晶型具有特征性。判断是否与已知晶型相同时,应该注意的是峰的相对位置(即2θ)而不是它们的相对强度。这是由于谱图(尤其在低角度)的相对强度会因为晶体条件、粒径或其它测定条件的差异产生的优势取向效果而变化,衍射峰的相对强度对于晶型的确定并非是特征性的。另外,同一个晶型的2θ值可能存在轻微误差,约为±0.2°。因此,在确定每种结晶结构时,应该将此误差考虑在内。在xrpd图谱中通常用2θ角或晶面距d值表示峰位置,两者之间具有简单的换算关系:d=λ/2sinθ,其中d值代表晶面间距,λ代表x射线的波长,θ为衍射角。还应特别指出的是,在混合物的鉴定中,由于含量下降等因素会造成部分衍射线缺失,此时,无需依赖高纯试样中观察到的全部谱带,一条谱带也可能对给定的晶体是特征性的。
dsc测定当晶体由于其晶体结构发生变化或晶体熔融而吸收或释放热时的转变温度。对于同种化合物的同种晶型,在连续的分析中,热转变温度和熔点误差典型的在约5℃之内。当我们说一个化合物具有一给定的dsc峰或熔点时,这是指该dsc峰或熔点±5℃。需要指出的是对于混合物而言,其dsc峰或熔点可能会在更大的范围内变动。此外,由于在物质熔化的过程中伴有分解,因此熔化温度与升温速率相关。
除非另外定义,本文使用的所有技术和科学术语具有与本发明所属领域的普通技术人员通常理解的相同的含义。
本发明化合物中的“氢”、“碳”、“氧”包括其所有同位素。同位素应理解为包括具有相同原子数但具有不同质量数的那些原子。举例来说,氢的同位素包括氕、氚和氘,碳的同位素包括13c和14c,氧的同位素包括16o和18o等。
附图说明
图1为式(i)化合物晶型a的x-射线衍射谱图;
图2为式(i)化合物晶型b的x-射线衍射谱图。
具体实施方式
下面代表性的实施例是为了更好地说明本发明,而非用于限制本发明的保护范围。
实施例1:(((1r,3s)-1-(5-((1s,2r)-1-氨基-2-羟丙基)-1,3,4-噁二唑-2-基)-3-(氰甲基)环丁基)氨基甲酰基)-l-丝氨酸的制备
步骤1:(4s,5r)-5-甲基-2-氧代噁唑烷酮-4-羧酸的制备
向l-苏氨酸(15.0g,125.92mmol)的氢氧化钠(1mol/l,450ml)溶液中缓慢滴加三(三氯甲基)碳酸酯(37.4g,125.92mmol)的1,4-二氧六环(300ml)溶液,滴完室温搅拌6小时,减压浓缩至干,加入热乙腈(450ml)搅拌,过滤,滤液浓缩至干,得标题化合物(18.1g,收率99.0%)。m/zcalcdforc5h7no4[m+h]+146.0,found146.1。
步骤2:2-((4s,5r)-5-甲基-2-氧代噁唑烷酮-4-羰基)肼-1-羧酸苄酯的制备
氮气保护下,-10℃下,向(4s,5r)-5-甲基-2-氧代噁唑烷酮-4-羧酸(10.0g,68.91mmol)的乙腈(250ml)溶液中依次滴加肼基甲酸苄酯(12.8g,25.8mmol)、三乙胺(52.3g,516.8mmol)和1-丙基磷酸酐(43.9g,137.8mmol)的乙酸乙酯溶液,滴完于-10℃下搅拌5小时,tlc监控反应终点。减压浓缩,加入水(250ml),乙酸乙酯(250mlx4)萃取,有机相干燥浓缩,所得残留物经柱层析色谱纯化,得标题化合物(5.8g,收率28.7%)。m/zcalcdforc13h15n3o5[m+h]+294.1,found294.1。
步骤3:(4s,5r)-5-甲基-2-氧代噁唑烷酮-4-碳酰肼的制备
向2-((4s,5r)-5-甲基-2-氧代噁唑烷酮-4-羰基)肼-1-羧酸苄酯(5.8g,19.8mmol)的甲醇(50ml)溶液中加入10%钯碳(0.6g),氢气置换,维持氢气氛围常压氢化,室温搅拌12小时,tlc监控反应终点,过滤,甲醇洗涤,有机相减压浓缩至干,得标题化合物(3.0g,收率95.3%)。m/zcalcdforc5h9n3o3[m+h]+160.06,found160.0。
步骤4:(1r,3r)-1-氨基-3-(羟甲基)环丁烷-1-羧酸甲酯的制备
氮气保护下,向(1r,3r)-1-氨基-3-(羟甲基)环丁烷-1-羧酸(500mg,3.44mmol)的甲醇(8ml)溶液中缓慢滴加二氯亚砜(491mg,4.13mmol),滴完回流2小时,tlc监控反应终点,减压浓缩至干,得标题化合物(550mg,81.7%)。m/zcalcdforc7h13no3[m+h]+160.01,found160.0。
步骤5:(1r,3r)-1-((叔丁氧羰基)氨基)-3-(羟甲基)环丁烷-1-羧酸甲酯的制备
向(1r,3r)-1-氨基-3-(羟甲基)环丁烷-1-羧酸甲酯(550mg,2.81mmol)的甲醇(11ml)溶液中依次加入二碳酸二叔丁酯(1.2g,5.62mmol)、三乙胺(2.8g,28.11mmol),50℃搅拌6小时,tlc监控反应终点,减压浓缩至干,所得残留物经柱层析色谱纯化,得标题化合物(510mg,收率70.1%)。m/zcalcdforc12h21no5[m-h]-258.1,found258.1。
步骤6:(1r,3r)-1-((叔丁氧羰基)氨基)-3-((甲磺酰氧基)甲基)环丁烷-1-羧酸甲酯的制备
氮气保护下,-10℃下向(1r,3r)-1-((叔丁氧羰基)氨基)-3-(羟甲基)环丁烷-1-羧酸甲酯(510mg,1.97mmol)的二氯甲烷(10ml)溶液中,依次滴入甲磺酰氯(338mg,2.95mmol)、三乙胺(399mg,3.94mmol),滴完室温搅拌1小时,减压浓缩,所得残留物经柱层析色谱纯化,得标题化合物(620mg,收率93.4%)。m/zcalcdforc13h23no7s[m-h]-336.12,found336.1。
步骤7:(1r,3r)-1-((叔丁氧羰基)氨基)-3-(氰甲基)环丁烷-1-羧酸甲酯的制备
氮气保护下,向(1r,3r)-1-((叔丁氧羰基)氨基)-3-((甲磺酰氧基)甲基)环丁烷-1-羧酸甲酯(100mg,0.30mmol)的n,n-二甲基甲酰胺溶液中依次加入碳酸钾(82mg,0.60mmol)、四丁基氟化铵(15μl,1mol/linthf)、三甲基氰硅烷(45mg,0.45mmol),80℃加热搅拌3小时,lc-ms监控反应终点,反应液倒入水中,二氯甲烷萃取,所得残留物经柱层析色谱纯化,得标题化合物(56mg,71%)。m/zcalcdforc13h20n2o4[m-h]-267.14,found267.1。
步骤8:(1r,3r)-1-((叔丁氧羰基)氨基)-3-(氰甲基)环丁烷-1-羧酸的制备
向(1r,3r)-1-((叔丁氧羰基)氨基)-3-(氰甲基)环丁烷-1-羧酸(5.0g,18.64mmol)的四氢呋喃/水(50ml,3:1)溶液中加入氢氧化锂(0.7g,27.95mmol),室温搅拌5小时,减压浓缩至干,加入水(50ml),二氯甲烷(20mlx2)萃取,水相用饱和柠檬酸调剂ph至3~4,乙酸乙酯(100mlx3)萃取,合并有机相,干燥,减压浓缩至干,得标题化合物(5.0g,收率94.8%)。m/zcalcdforc12h18n2o4[m-h]-253.13,found253.1。
步骤9:((1r,3r)-3-(氰甲基)-1-(2-((4s,5r)-5-甲基-2-氧代噁唑烷酮-4-羰基)肼-1-羰基)环丁基)氨基甲酸叔丁酯的制备
氮气保护下,向(1r,3r)-1-((叔丁氧羰基)氨基)-3-(氰甲基)环丁烷-1-羧酸(3.8g,14.90mmol)的二氯甲烷(50ml)溶液中依次加入(4s,5r)-5-甲基-2-氧代噁唑烷酮-4-碳酰肼(2.9g,17.89mmol)和三乙胺(7.5g,74.50mmol)。控制温度低于-15℃以下缓慢滴入1-丙基磷酸酐(9.5g,29.8mmol)的乙酸乙酯溶液。滴完后-10℃下反应5小时,tlc监控反应终点。加入水(50ml)搅拌20分钟,柠檬酸饱和溶液调节ph至4~5,乙酸乙酯(100mlx5)萃取,有机相浓缩,所得残留物经柱层析色谱纯化,得标题化合物(5.5g,收率97.8%)。m/zcalcdforc17h25n5o6[m-h]-394.18,found394.1。
步骤10:((1r,3s)-3-(氰甲基)-1-(5-((4s,5r)-5-甲基-2-氧代噁唑烷酮-4-基)-1,3,4-噁二唑-2-基)环丁基)氰基甲酸叔丁酯的制备
氮气保护下,向((1r,3r)-3-(氰甲基)-1-(2-((4s,5r)-5-甲基-2-氧代噁唑烷酮-4-羰基)肼-1-羰基)环丁基)氨基甲酸叔丁酯(2.4g,6.12mmol)的乙腈(100ml)溶液中加入4-甲基苯磺酰氯(2.3mmol,12.24mmol),室温下缓慢滴入n,n-二异丙基乙胺(2.4g,18.4mmol),滴完后升温至45℃反应5小时,tlc监控反应终点。反应结束后恢复至室温,乙酸乙酯(200mlx3)萃取,有机相减压浓缩,所得残留物经柱层析色谱纯化,得标题化合物(1.2g,收率52.0%)。m/zcalcdforc17h23n5o5[m-h]-376.17,found376.2。
步骤11:2-((1s,3r)-3-氨基-3-(5-((4s,5r)-5-甲基-2-氧代噁唑烷酮-4-基)-1,3,4-噁二唑-2-基)环丁基)乙腈三氟乙酸盐的制备
氮气保护下,向((1r,3s)-3-(氰甲基)-1-(5-((4s,5r)-5-甲基-2-氧代噁唑烷酮-4-基)-1,3,4-噁二唑-2-基)环丁基)氰基甲酸叔丁酯(1.5g,3.97mmol)的二氯甲烷(15ml)溶液中滴加三氟乙酸(4.5g,39.70mmol),滴完室温搅拌2h,tlc监控反应终点。减压浓缩至干,所得残留物经柱层析色谱纯化,得标题化合物(1.2g,77.3%)。1hnmr(d2o,400mhz)δ5.02~5.07(m,2h),3.03~3.13(m,1h),2.76~2.77(d,j=4hz,2h),2.59~2.67(m,4h),1.59~1.61(d,j=8hz,3h);m/zcalcdforc12h15n5o3[m+h]+278.12,found278.2。
步骤12:(((1r,3s)-3-(氰甲基)-1-(5-((4s,5r)-5-甲基-2-氧代噁唑烷酮-4-基)-1,3,4-噁二唑-2-基)环丁基)氨基甲酰基)-l-丝氨酸甲酯的制备
氮气保护下,在-20℃下,向四氢呋喃(5ml)溶液中依次缓慢滴入三光气(272mg,0.92mmol)、o-(叔丁基二甲硅基)-l-丝氨酸甲酯(143mg,0.61mmol)的四氢呋喃(5ml)溶液;滴完,室温搅拌2小时,tlc监控反应终点。减压浓缩至干,用四氢呋喃(5ml)溶解,加入三乙胺(263mg,2.60mmol),将2-((1s,3r)-3-氨基-3-(5-((4s,5r)-5-甲基-2-氧代噁唑烷酮-4-基)-1,3,4-噁二唑-2-基)环丁基)乙腈三氟乙酸盐(200mg,0.51mmol)的四氢呋喃(5ml)溶液室温下滴入,滴完室温搅拌12h,减压浓缩,二氯甲烷(50ml)溶解,饱和柠檬酸溶液洗涤,有机相饱和食盐水洗涤,加入柱层析硅胶(2g)搅拌5h,过滤,有机相减压浓缩,所得残留物经柱层析色谱纯化,得标题化合物(120mg,收率55.8%)。m/zcalcdforc17h22n6o7[m+h]+423.15,found423.2。
步骤13:(4s,5r)-4-(5-((1r,3s)-3-(氰甲基)-1-(3-((s)-3-羟基-1-甲氧基-1-氧丙烷-2-基)脲)环丁基)-1,3,4-噁二唑-2-基)-5-甲基-2-氧代噁唑烷酮-3-羧酸叔丁酯的制备
氮气保护下,向(((1r,3s)-3-(氰甲基)-1-(5-((4s,5r)-5-甲基-2-氧代噁唑烷酮-4-基)-1,3,4-噁二唑-2-基)环丁基)氨基甲酰基)-l-丝氨酸甲酯(100mg,0.24mmol)的乙腈(5ml)溶液中依次滴加二碳酸二叔丁酯(78mg,0.36mmol)的乙腈(2ml)溶液、4-二甲氨基吡啶(15mg,0.12mmol)的乙腈(2ml)溶液。滴完室温搅拌4h,tlc监控反应终点。减压浓缩,二氯甲烷溶解,水洗,饱和食盐水洗涤,无水硫酸钠干燥,减压浓缩,所得残留物经柱层析色谱纯化,得标题化合物(75mg,59.9%)。m/zcalcdforc22h30n6o9[m-h]-521.21,found521.2。
步骤14:(((1r,3s)-1-(5-((1s,2r)-1-((n-叔丁氧羰基)氨基)-2-羟丙基)-1,3,4-噁二唑-2-基)-3-(氰甲基)环丁基)氨基甲酰基)-l-丝氨酸甲酯的制备
氮气保护下,向(4s,5r)-4-(5-((1r,3s)-3-(氰甲基)-1-(3-((s)-3-羟基-1-甲氧基-1-氧丙烷-2-基)脲)环丁基)-1,3,4-噁二唑-2-基)-5-甲基-2-氧代噁唑烷酮-3-羧酸叔丁酯(200mg,0.38mmol)的甲醇溶液中加入碳酸铯(124mg,0.38mmol),室温搅拌4h,减压浓缩,所得残渣经柱层析色谱纯化,得标题化合物(150mg,79.6%)。m/zcalcdforc21h32n6o8[m-h]-495.23,found495.2。
步骤15:(((1r,3s)-1-(5-((1s,2r)-1-氨基-2-羟丙基)-1,3,4-噁二唑-2-基)-3-(氰甲基)环丁基)氨基甲酰基)-l-丝氨酸的制备
氮气保护下,向(((1r,3s)-1-(5-((1s,2r)-1-((n-叔丁氧羰基)氨基)-2-羟丙基)-1,3,4-噁二唑-2-基)-3-(氰甲基)环丁基)氨基甲酰基)-l-丝氨酸甲酯(500mg,1.01mmol)的四氢呋喃/水(3:1,10ml)溶液中加入氢氧化锂(36mg,1.52mmol),室温搅拌5h,tlc监控反应终点,减压浓缩至干,加入水(20ml),二氯甲烷(10mlx2)萃取,水相用饱和柠檬酸溶液调ph至3~4,乙酸乙酯(20mlx3)萃取,有机相无水硫酸钠干燥,减压浓缩至干,依次加入二氯甲烷(5ml)、三氟乙酸/苯酚(5ml,95:5),氮气保护下室温搅拌0.5h,hplc监控反应终点,室温减压浓缩,低温减压干燥30min,将反应液倒入20ml甲基叔丁基醚中,-15℃下搅拌1h,过滤,所得粗品用乙醇(5ml)溶解,氨水调节ph至5~6,析出白色固体,滴加异丙醇(5ml),室温搅拌1h,过滤,干燥得标题化合物(289mg,74.9%)。1hnmr(d2o,400mhz)δ4.70~4.72(d,j=8hz,1h),4.33~4.40(m,1h),4.10~4.12(t,j=4hz,1h),3.63~3.69(m,2h),3.02~3.09(m,1h),2.58~2.78(m,6h),1.32~1.34(d,j=8hz,3h);m/zcalcdforc15h22n6o6[m+h]+383.16,found383.2。
实施例2(((1r,3s)-1-(5-((1s,2r)-1-氨基-2-羟丙基)-1,3,4-噁二唑-2-基)-3-(氰甲基)环丁基)氨基甲酰基)-l-丝氨酸晶型a的制备
取实施例1中制备的(((1r,3s)-1-(5-((1s,2r)-1-氨基-2-羟丙基)-1,3,4-噁二唑-2-基)-3-(氰甲基)环丁基)氨基甲酰基)-l-丝氨酸(100mg)于25ml单口烧瓶中,加入甲醇(15ml),回流状态下至溶清,回流20分钟后关闭加热自然冷却降温12h,过滤,干燥,收集样品进行xrpd表征,表征结果显示本次实验晶型为晶型a,xrpd图谱见图1。
实施例3(((1r,3s)-1-(5-((1s,2r)-1-氨基-2-羟丙基)-1,3,4-噁二唑-2-基)-3-(氰甲基)环丁基)氨基甲酰基)-l-丝氨酸晶型b的制备
取实施例1中制备的(((1r,3s)-1-(5-((1s,2r)-1-氨基-2-羟丙基)-1,3,4-噁二唑-2-基)-3-(氰甲基)环丁基)氨基甲酰基)-l-丝氨酸(50mg)于25ml单口烧瓶中,加入5ml乙醇,25±5℃下搅拌2h,过滤,干燥,收集样品进行xrpd表征,表征结果显示本次实验晶型为晶型b,xrpd图谱见图2。
比较例1
根据wo2015/033301(pct/ib2014/064281)中实施例2公开的方法制备下式代表的化合物(化合物a),并通过氢谱和质谱鉴定,
使用以下实验例1和2的方法测试了化合物a的药代动力学特征以及在结肠癌ct26细胞皮下移植瘤模型上的抑瘤效果,实验结果显示,化合物a的生物利用度(f)和抑瘤率弱于本发明的化合物。
另外,本发明的发明人还根据wo2015/033301公开的方法合成并测试了wo2015/033301表3中compoundno.12,结果显示,compoundno.12的生物利用度和抑瘤率明显弱于本发明的化合物及以上的化合物a。
实验例1药物代谢实验
1实验材料
1.1化合物
使用以上实施例制备的本发明的化合物和wo2015/033301中实施例2的化合物(“化合物a”)进行该实验。口服药物用生理盐水溶解,制成0.5mg/ml澄清溶液,静脉药物用生理盐水溶解,制成0.1mg/ml澄清溶液。
1.2动物
雄性balb/c小鼠,每组各3只,体重18-22g,上海西普尔-必凯实验动物有限公司提供。
受试小鼠实验前给予2~4天的环境适应期,给药前禁食8-12h,给药2h后给水,4h后给食。
1.3试剂
甲醇(色谱纯):spectrum公司生产;
乙腈(色谱纯):spectrum公司生产;
其余试剂均为市售分析纯。
1.4仪器
美国ab公司api4500型三重四级杆液质联用仪,配有电喷雾离子源(esi),lc-30ad双泵;sil-30ac自动进样器;cto-30ac柱温箱;dgu-20a3r脱气机;analystqsa01.01色谱工作站;milli-q超纯水器(milliporeinc);qilinbeiervortex-5振荡器;hitachicf16rⅹⅱ台式高速冷冻离心机。
2实验方法
1)小鼠禁食但可自由饮水12小时后,采取0时刻空白血浆;
2)取步骤1)中的小鼠,灌胃(intragastricadministration,ig)给予待测化合物10mg/kg;静脉(iv)给予待测化合物1mg/kg;
3)于灌胃后5min,15min,30min,1h,2h,4h,8h,10h,24h,从眼底静脉丛连续取血置于分布有肝素的ep管中,8000rpm/min离心5min后取上层血浆,-20℃冻存,待lc-ms/ms分析;
4)根据步骤3)所得的血药浓度-时间数据,采用winnonlin软件求算药代动力学参数;
3实验结果:
药代动力学实验数据如表1中所示,结果表明口服给予小鼠实施例1的化合物,具有非常好的半衰期、曲线下面积以及生物利用度,成药性好,具有良好的临床应用前景。
表1本发明实施例化合物的药代动力学数据
实验例2体内药效实验
1、实验材料
1.1化合物
使用根据本发明实施例制备的化合物进行该实验。阴性对照组给予生理盐水。采用口服给药,测试化合物用生理盐水溶解,制成2mg/ml澄清溶液。
1.2动物
雌性balb/c小鼠,每组各3只,体重18-22g,上海西普尔-必凯实验动物有限公司提供。受试小鼠实验前给予2~4天的环境适应期,给药前禁食8-12h,给药2h后给水,4h后给食。
1.3阳性对照
化合物a,根据wo2015/033301实施例2公开的方法制备。
2、实验方法
接种细胞后待肿瘤生长至平均体积为50mm3后,将动物随机分组,每组6只,各测试组口服给药20mg/kg,一天一次,连续给药14天。考察实验动物体重的变化及肿瘤生长是否被抑制或延缓。每周三次用游标卡尺测量肿瘤直径。
肿瘤体积的计算公式为:v=0.5a×b2,a和b分别表示肿瘤的长径和短径。
相对肿瘤增殖率t/c(%)的计算公式为:t/c=trtv/crtvx100%(trtv:治疗组rtv;crtv:溶剂对照组rtv)。根据测量结果计算出相对肿瘤体积(relativetumorvolume,rtv),rtv=vt/v0,其中v0为实验开始时的肿瘤体积,vt每一次测量时的肿瘤体积。
肿瘤生长抑制率tgi(%)的计算公式为:tgi=[1-(某处理组给药结束时平均瘤体积-该处理组开始给药时平均瘤体积)/(溶剂对照组治疗结束时平均瘤体积-溶剂对照组开始治疗时平均瘤体积)]x100%。
3、实验结论
3.1体重变化情况
本发明的化合物对小鼠结肠癌ct26细胞皮下同系移植瘤在balb/c小鼠模型的体重无影响,表2给出了实施例1以及化合物a给药后对体重的影响,表明对照组及给药组动物体重在给药期间逐渐增加,具有较好的耐受性。
表2本发明的化合物给药后对小鼠体重的影响
3.2抗肿瘤药效评价指标
药效评价指标如表3所示,给药后第14天时溶剂对照组荷瘤鼠的平均瘤体积达到1524mm3,实施例1的化合物在第14天时的t/c值为39.0%,tgi值为63.7%,表示其对ct26结肠癌细胞移植瘤具有显著的抑制作用。化合物a组在第14天时的t/c值为63.9%,tgi值为37.8%,抑瘤率明显低于本发明的化合物。
表3抗肿瘤药效评价指标
注:a:均值±sem;b:肿瘤生长抑制,由t/c和tgi表示;c:p值根据肿瘤体积计算
尽管以上已经对本发明作了详细描述,但是本领域技术人员理解,在不偏离本发明的精神和范围的前提下可以对本发明进行各种修改和改变。本发明的权利范围并不限于上文所作的详细描述,而应归属于权利要求书。
- 还没有人留言评论。精彩留言会获得点赞!