一种C70-BODIPY分子及其制备方法与流程
本发明涉及功能有机分子设计与合成技术领域,具体是涉及一种c70-bodipy分子及其制备方法。
背景技术:
三重态光敏化剂是一种广泛应用在三重态-三重态湮灭上转换、光催化有机反应、光动力学疗法等领域的复合材料。由于含富勒烯(主要是c60)的三重态光敏化体系具有稳定的系间窜越性质,近年来成为研究的热点[huang,d.;zhao,j.;wu,w.;yi,x.;yang,p.;ma,j.asianj.org.chem.2012,1,264;huang,l.;cui,x.;therrien,b.;zhao,j.chem.eur.j.2013,19,17472;huang,l.;zhao,j.chem.commun.2013,49,3751;wu,w.;zhao,j.;sun,j.;guos.j.org.chem.2012,77,5305;huang,l.;yu,x.;wu,w.;zhao,j.org.lett.,2012,14,2594;zhu,s.-e;liu,k.-q.;wang,x.-f.;xia,a.-d.;wang,g.-w.j.org.chem.,2016,81,12223.]。
c70也是比较稳定的富勒烯分子,而且其含量仅次于c60,约占c60含量的1/4。c70分子具有d5h对称性,由于分子的对称性不如c60好,具有更强的s0-s1吸收,因此c70对可见光的吸收比c60强[hung,r.r.;grabowski,j.j.chem.phys.lett.,1992,192,249]。与c60相比,c70是更好的电子接受体[xu,w.;feng,l.;wu,y.;wang,t.;wu,j.;xiang,j.;li,b.;jiang,l.;shuc.;wang,c.phys.chem.chem.phys.,2011,13,428]。kim等发现c70作为敏化剂应用在三重态-三重态上转换中,其转换效率比c60高25~35倍[moor,k.;kim,j.-h.;snow,s.;kim,j.-h.chem.commun.,2013,49,10829]。因此,与c60相比,c70有望成为更加理想的自旋转换单元。
但是,到目前为止,关于c70的三重态光敏化体系的报道还比较少[wei,y.;zheng,m.zhou,q.;zhou,x.;lius.]。而且作者将bodipy连接到c70吡咯环的“2”位,bodipy分子与c70呈现一定的夹角。在连接方式上,本发明将bodipy通过刚性桥基连接在c70吡咯环的“n”位上,形成c2v对称的c70-bodipy分子。这样bodipy、碳桥及c70在一条直线上,确保能量或电荷转移都在一条直线上完成,简化体系的光物理过程,也排除了其它因素对光敏化效率的影响。
技术实现要素:
本发明的目的之一在于提供一种c70-bodipy分子,它既能够克服其它分子系间窜越性质不稳定的缺点,拓宽三重态光敏化剂的类型,又可以简化体系的光物理过程。
为实现该目的,本发明采用了以下技术方案:
一种c70-bodipy分子,其分子结构通式如下所示:
通式中r1和r2选自取代的芳基或杂环芳基、碳原子数为2~5的烷基、h、cl、br、i、-or或sr,其中r选自h、取代的芳基或杂环芳基、碳原子数为2~6的烷基;
通式中r3选自碳原子数为1~12的烷基;
通式中r4选自h、cl、br、i、-nr2’或sr’、-or’、
通式中x为
上式中bodipy通过桥基连接在c70吡咯环“n”位上,形成具有c2v对称的c70-bodipy分子。
本发明的另一目的在于提供一种c70-bodipy分子的制备方法,为实现该目的,本发明采用了以下技术方案:
制备路线为:
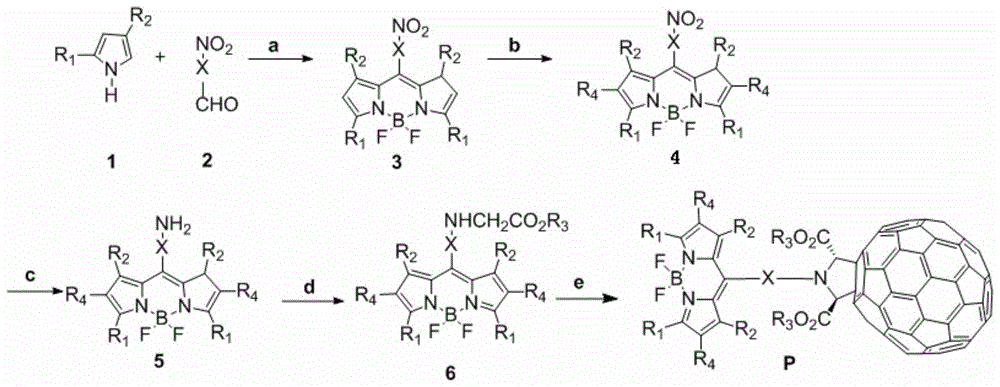
依次包括如下步骤:
步骤a:2,4-位取代的吡咯1与硝基取代的醛2在二氯甲烷或氯仿中在路易斯酸(如三氟乙酸或三氟化硼乙醚等)的催化下发生缩合反应,然后再用氧化剂(如ddq,邻四氯苯醌,四氯对苯醌)进行氧化脱氢,再用碱中和后,用三氟化硼乙醚(bf3·oet2)配位,制备含硝基的bodipy分子3;其中摩尔投料比1:2:酸:氧化剂:bf3·oet2=1.5~2.5:0.8~1.2:0.15~0.25:0.8~1.2:30~60,反应温度为0~30℃,反应时间为36~72h;
步骤b:当r4=br或i时,化合物4的制备方法是:化合物3与n-溴或碘代丁二酰亚胺在酸(如乙酸)的催化下,在有机溶剂(如氯仿、二氯甲烷等)中反应生成化合物4,其中摩尔投料比3:n-溴或碘代丁二酰亚胺=0.8~1.2:2.4~6,反应温度为0~60℃,反应时间为8~24h;
当r4=-nr2’、sr’或-or’时,化合物4的制备方法是:化合物3与n-碘代丁二酰亚胺在酸(如乙酸)的催化下,在有机溶剂(如氯仿、二氯甲烷等)中反应生成吡咯环上被碘取代的中间体,该中间体再与hnr2’或hsr’或hor’在强碱(如naoh、nah等)存在下,在极性溶剂(如四氢呋喃、dmf、丙酮等)中发生亲核取代反应,合成化合物4,其中摩尔投料比3:n-碘代丁二酰亚胺=0.8~1.2:2.4~6,反应温度为0~60℃,反应时间为8~24h;摩尔投料比被碘取代的中间体:hnr2’或hsr’或hor’:强碱=0.8~1.2:2~4.8:2~4.8,反应温度为25~60℃,反应时间为8~36h;
当
当r4=h时,跳过步骤b;
步骤c:化合物4在溶剂(如四氢呋喃等醚类)中利用兰尼镍的催化,选用nh2nh2·h2o还原制备氨基bodipy化合物5,化合物4、nh2nh2·h2o与兰尼镍的比例为0.8mmol~1.2mmol:2.5mmol~3.6mmol:0.33g~0.5g,反应温度为40~80℃,反应为时间2~10h;
步骤d:化合物5在无水碳酸钾和三乙胺的催化作用下与溴乙酸烷基酯发生取代反应制备产物6,其中化合物5、无水碳酸钾、三乙胺与溴乙酸烷基酯的摩尔比为0.8~1.2:0.8~2.4:4~12:0.8~1.5,反应温度为20~60℃,反应时间为10~48h;
步骤e:化合物6、c70与乙醛酸烷基酯通过1,3-偶极环加成反应制备目标产物p,其中化合物6、c70与乙醛酸烷基酯的摩尔比为0.8~3.6:0.8~1.2:8~24,反应温度为110~170℃,反应时间为4~10h。
作为本发明的c70-bodipy分子的制备方法的进一步改进:
所述步骤a具体操作为:将化合物2以1:1000~1500(g:ml)溶解在无水二氯甲烷中,再缓慢加入化合物1;混合液在室温下搅拌,并且剧烈地通惰性气体20~40min后,滴加2滴三氟化硼乙醚;反应混合液继续在惰性气体环境中,在室温下反应12~24h,直到tlc板显示化合物2消失为止;将1倍当量左右的氧化剂(如ddq,邻四氯苯醌,四氯对苯醌)缓慢地加到上述反应混合液中;继续搅拌4~10h后,反应液转移到冰浴条件下;用恒压滴液漏斗慢慢地加入三乙胺(et3n),搅拌10~30min后,再缓慢地加入三氟化硼乙醚(bf3·oet2);混合物在室温下继续搅拌4~12h后,停止反应;反应液用有机溶剂(如二氯甲烷、氯仿、乙酸乙酯等)和水反复萃取三次后,用无水硫酸钠干燥过夜;旋干溶剂后,用柱色谱分离,得到橘黄色固体的产物3。
所述步骤b具体操作为:
当r4=br或i时,制备化合物4的具体操作是:将化合物3以1:50~100(g:ml)溶解在氯仿和乙酸的混合溶液中(两种溶剂的比例为2~4:1),再加入n-溴或碘代丁二酰亚胺,混合液在室温下搅拌,待反应完成后,加入硫代硫酸钠的水溶液淬灭反应;反应混合液再用碳酸钠水溶液和氯仿萃取,干燥后,用柱色谱分离,即可得到化合物4;
当r4=-nr2’、sr’或-or’时,制备化合物4的具体操作是:将碘代中间体以1:30~80(g:ml)溶解在极性溶剂(如四氢呋喃、dmf、丙酮等)中,再加入nah,然后再加入hnr2’或hsr’或hor’,反应混合物在室温下反应,待中间体基本消失后,加入水淬灭反应,用氯仿萃取,经过干燥和柱色谱分离后,得到化合物4;
当
所述步骤c具体操作为:将化合物4以1:10~30(g:ml)溶解在溶剂(如四氢呋喃等醚类)中,然后加入水合肼和兰尼镍;直到点板确认化合物4消失为止,停止反应;过滤除去兰尼镍后,除去溶剂,过柱分离得到化合物5。
所述步骤d具体操作为:取化合物5以1:25~100(g:ml)溶解于丙酮中,然后再加三乙胺和无水碳酸钾;反应混合物在室温下先通氩气保护15~30min,然后迅速地加入溴乙酸烷基酯(brch2co2r),继续通5~10min氩气;反应混合物放在油浴锅进行反应,利用薄层色谱跟踪,待原料点基本消失,除去丙酮,再将混合物过柱分离,得到化合物6。
所述步骤e具体操作为:将c70以1:40~50(g:ml)溶解在溶剂(如邻二氯苯、氯苯或甲苯)中,并加入化合物6;反应混合物在室温下,通氩气保护15~30min,然后将乙醛酸烷基酯(hcoco2r)加入,继续通10~15min氩气,将反应混合物放入预先加热的油浴锅中反应;待富勒烯的量剩余20%~30%时,停止反应;然后减压蒸馏,将反应体系中的溶剂除去;反应混合物用二硫化碳或甲苯溶解,并用柱色谱分离,先以二硫化碳或甲苯作为洗脱剂,去除未反应完的c70,然后再改用大极性的淋洗剂,得到产物p。
本发明提出的一种c70-bodipy分子,其结构通式中bodipy分子的meso位置与富勒烯吡咯环结构的n原子相连,由于c70的对称性较差,因此形成三种不同位点加成的目标产物。与现有技术相比,本发明的有益效果表现在:
1)本发明利用结构独特的c70作为自旋转换单元,通过合理设计与bodipy连接在一起,形成具有稳定系间窜越性质的分子,不仅克服其它分子系间窜越性质不稳定的缺点,而且拓展了自旋转换单元的种类。
2)本发明制备的c70-bodipy分子能够产生较强的光敏化效果。
3)本发明的c70-bodipy分子可应用在光催化有机反应、光动力学疗法(pdt)、润滑油抗氧化、金属加工液的杀菌等领域。
附图说明
图1是c70-bodipy分子的制备路线图。
图2是实施例制备的c70-bodipy分子p1的1hnmr。
图3是实施例的c70-bodipy分子p1的13cnmr。
图4是实施例的c70-bodipy分子p1的质谱。
具体实施方式
本实施例以一种c70-bodipy分子p1为例,结合制备路线图(图1所示)介绍其结构以及具体的制备方法。
中间体3-1的制备方法:
称取2,4-二甲基吡咯0.7221g,转移至1000ml的三口圆底烧瓶中,取对硝基苯甲醛0.4616g,然后放入搅拌磁子。再用量筒量取500ml的无水二氯甲烷将原料溶解,在室温下匀速搅拌并通入氮气30min。然后快速打开三口烧瓶中的一口,并用胶头滴管加入1-2滴三氟化硼乙醚。在25℃条件下(室温),继续反应并搅拌12h。然后tlc跟踪,当板上显示原料点对硝基苯甲醛完全消失,则停止通氮气。
然后称取0.7330g的邻四氯苯醌,用10ml的二氯甲烷将其溶解,在三口烧瓶中间口用恒压滴液漏斗缓慢加入。滴加完后,继续在室温下搅拌并反应4h,并将三口烧瓶的三个口用真空塞封上。然后将三口烧瓶放入冰水中,用量筒量取18ml的三乙胺用恒压滴液漏斗于三口烧瓶中间缓慢加入,且三口烧瓶的左右口敞开,滴完后继续搅拌30min。再用量筒量取19ml的三氟化硼乙醚用恒压滴液漏斗于三口烧瓶中间缓慢滴加,注意:三口烧瓶的左右口还要敞开。滴加完毕后移除冰水,改为室温条件下,反应并搅拌12h后,结束本次反应。萃取反应液:加入反应液、二氯甲烷和水之后,摇晃2-3次分液漏斗,放在铁架台上静置分层,反应液与二氯甲烷的混合相在分液漏斗下层,反复萃取反应液和水相2-3次,最后用无水硫酸钠干燥萃取后的反应液半天。将其过滤得到除去水分的反应液,然后用旋转蒸发仪将反应液旋干得到黑绿色的油状物。再用少量二氯甲烷溶解产物,用石油醚:二氯甲烷=1:1过柱分离。干燥后得到0.28g的橘黄色固体产物3-1。
中间体5-1的制备方法:
将531mg的化合物3-1溶解在20ml四氢呋喃中,并通入氮气搅拌30min来除去反应体系中的氧气。然后加入0.6ml的水合肼和98.5mg的兰尼镍。将该三口烧瓶放于磁力搅拌器中油浴加热回流。用tlc跟踪,反应6h后,待3-1全部消失时,停止反应。当反应液恢复至室温时,过滤除去兰尼镍后,除去溶剂。再用少量二氯甲烷溶解产物,石油醚:二氯甲烷=1:1作为淋洗剂过柱分离,得到430mg的化合物5-1。
化合物6-1的合成步骤:
称取0.2059g的化合物5-1溶解于20ml的丙酮中,然后再加入2ml的三乙胺和0.1503g的k2co3;反应混合物在室温下先通氩气保护20min,然后加入68μl的溴代乙酸乙酯(brch2co2et),继续通5~10min氩气;通完氮气后快速用橡皮塞封住单口烧瓶,再裹上一层保鲜膜,绑上橡皮筋。将单口烧瓶放于磁力搅拌器中油浴加热搅拌8h,油浴锅温度为30℃。用tlc爬板,将石油醚:乙酸乙酯=5:1作为展剂,直至5-1基本消失完全,停止反应。等待反应液恢复至室温,然后用旋转蒸发仪将反应液旋干,除去丙酮,再将混合物过柱分离,得到65.11mg的黄色化合物6-1。
化合物p-1的合成步骤:
将250.4mg的化合物6-1和50mg的c70溶解于7ml的邻二氯苯中。反应混合物在室温下,通氩气保护30min,然后往反应液里加入150μl的乙醛酸乙酯,继续通10min氮气,然后将反应液在150℃的油浴下反应,用tlc点板跟踪,直到c70剩下20%~30%时停止反应。将反应液恢复至室温,然后减压除去反应液中的邻二氯苯。然后用二硫化碳溶解产物,先用纯二硫化碳作为淋洗剂,分离出未反应的c70,然后换淋洗剂为二硫化碳:二氯甲烷=1:1,最后干燥得到16mg目标产物p-1。
上述实施例制备的目标产物p-1的1hnmr,13cnmr分别如图2、3所示,实验测试该分子的质谱数据:m/z=1349.6511(c97h30bf2n3o4,m+)与预测分子量吻合。(如图4所示)。
上述制备的目标产物p-1的结构式如下所示:
对于本领域技术人员来说,随着目标产物p(结构通式如下)通式中r、x的不同选择,将会制备出若干种不同结构的c70-bodipy分子,分子结构通式为:
示例1(c70-bodipy分子p-2):
通式中,r1,r2为甲基,r3为乙基,r4为i,x为
示例2(c70-bodipy分子p-3):
通式中,r1,r2为乙基,r3为乙基,r4为乙基,x为
示例3(c70-bodipy分子p-4):
通式中,r1,r2为甲基,r3为正丁基,r4为己基,x为丁基。
示例4(c70-bodipy分子p-5):
通式中,r1,r2为乙基,r3为十二烷基,r4为丙基,x为
示例5(c70-bodipy分子p-6):
通式中,r1,r2为甲基,r3为戊基,r4为咔唑基,x为
示例6(c70-bodipy分子p-7):
通式中,r1,r2为甲基,r3为乙基,r4为
上述不同的c70-bodipy分子(p-2~p-7),其制备方法及反应机理与p-1较为雷同,在此不再一一赘述。
- 还没有人留言评论。精彩留言会获得点赞!