一种硫酸氢氯吡格雷晶型II的制备方法与流程
本发明涉及一种硫酸氢氯吡格雷晶型ii的制备方法,属于药物化学结晶技术领域。
背景技术:
硫酸氢氯吡格雷是由法国sanlfi-aventis公司于1986年研发的抗血小板凝聚类药物,它通过选择性的与血小板表面腺苷酸环化酶偶联的adp受体结合而不可逆地抑制血小板聚集,减少血管中血栓形成。该药临床疗效显著、副作用少、耐受性高,逐渐替代阿司匹林定,已成为治疗血栓性疾病的一线药物。该产品1998年首次在美国和英国上市,于2001年进入中国。目前,国内硫酸氢氯吡格雷制剂产品的主要生产商有赛诺菲(杭州)制药有限公司、深圳信立泰药业股份有限公司、乐普药业股份有限公司。
现有的文献中制备硫酸氢氯吡格雷的方法有很多,大多数反应较为复杂,反应条件苛刻,不利于大批量的生产。其中,中国专利cn104370935b涉及一种硫酸氢氯吡格雷的制备方法,它以2-噻吩乙醛为基本起始原料,使其与邻氯苯甘氨酸甲酯发生缩合反应,生成相应的亚胺中间体,再用硼氢化钠或直接用腈基硼氢化钠将亚胺中间体还原为相应的仲胺中间体,然后这种仲胺中间体与甲醛反应关环得到氯吡格雷,经硫酸酸化后得到目标化合物硫酸氢氯吡格雷。
中国专利申请cn107383055a涉及一种硫酸氢氯吡格雷的合成方法,以邻苯氯乙酸和4,5,6,7四氢噻吩[3,2-c]为原料,包括如下步骤:(1)制备酰化反应;(2)溴化反应;(3)酯化反应;(4)制备氯吡格雷碱粗品;(5)氯吡格雷碱提纯;(6)消旋体拆分;(7)制备硫酸氢氯吡格雷。
国际专利申请wo2003/051362报道了一种以丙酮为溶剂制备硫酸氢氯吡格雷方法,此方法需要低温,不利于节能降耗。
中国专利cn101643476b报道一种硫酸氢氯吡格雷晶型ii的制备方法,将硫酸氢氯吡格雷晶型i溶解于低级醇,然后加入反溶剂得到ii晶型,此法繁琐,成品收率低,且耗费的工时多,能耗高,不利于降低成本。
再次,专利报道的一些合成方法,在有关杂质含量方面也不是很理想。美国usp标准中规定了需要控制氯吡格雷三个已知杂质,即杂质a(casno.:144750-42-5),杂质b(casno.:144750-52-7),杂质c(casno.:120202-71-3)。其中a是氯吡格雷的水解产物,b是同分异构体,杂质c主要是右旋硫酸氢氯吡格雷的光学对映体。fda规定这三种杂质不得超过hplc积分面积的0.1%,主要是由于三种有关杂质a,b,c均对硫酸氢氯吡格雷的药效有一定影响,因此需要对其含量加以控制。
鉴于目前制备硫酸氢氯吡格雷的工艺复杂,反应条件苛刻,不利于大生产,且硫酸氢氯吡格雷晶型ii的制备方法得到的产品杂质含量偏高的现状,亟需开发一种新的制备方法,能够采用简单易得的物料,用较为温和的反应条件,得到高纯度的硫酸氢氯吡格雷晶型ii。
技术实现要素:
本发明提供了一种硫酸氢氯吡格雷晶型ii的制备方法,旨在解决硫酸氢氯吡格雷的工艺复杂,反应条件苛刻,且杂质含量偏高的问题,降低成本投入,一次性制备出一种高纯度的硫酸氢氯吡格雷晶ii型,反应过程安全环保。
本发明涉及一种硫酸氢氯吡格雷晶型ii的制备方法,其包括以下步骤:
s1.(+)邻氯苯甘氨酸甲酯酒石酸盐通过水解反应获得(+)邻氯苯甘氨酸甲酯;
s2.加入乙腈,无机碱,与所述(+)邻氯苯甘氨酸甲酯混合,20~30℃搅拌;再加入2-(2-噻吩基)乙醇对甲苯磺酸酯,然后升温至48~52℃,保温反应,反应完后离心,将母液调至ph为3~5,搅拌,离心,所得固体在50~60℃干燥,得到(+)α-(2-噻吩乙胺基)-α-(2-氯苯基)乙酸甲酯盐酸盐;
s3.将所述(+)α-(2-噻吩乙胺基)-α-(2-氯苯基)乙酸甲酯盐酸盐与甲醛混合,加热,保温反应完全。然后冷却降温,加入无机碱、水和二氯甲烷,搅拌分液,萃取,合并有机层,获得(+)氯吡格雷游离碱;
s4.将所述(+)氯吡格雷游离碱消旋拆分,获得(+)氯吡格雷樟脑磺酸复盐;
s5.将所述(+)氯吡格雷樟脑磺酸复盐水解获得(+)氯吡格雷游离碱i;
s6.将所述(+)氯吡格雷游离碱i与丙酮混合,过滤,滤液降温加入等摩尔的浓硫酸,搅拌,离心,干燥,得到硫酸氢氯吡格雷晶型ii。
根据本发明所述硫酸氢氯吡格雷晶型ii的制备方法,其中s1步骤中,将萃取剂,水,无机碱,搅拌溶清,再加入(+)邻氯苯甘氨酸甲酯酒石酸盐,反应完全后分液,水层用所述萃取剂萃取,合并有机层,用干燥剂干燥,离心,取滤液减压回收所述萃取剂,得(+)邻氯苯甘氨酸甲酯。
根据本发明所述硫酸氢氯吡格雷晶型ii的制备方法,其中s2步骤中,所述无机碱与所述(+)邻氯苯甘氨酸甲酯的摩尔比为2~2.5:1。
根据本发明所述硫酸氢氯吡格雷晶型ii的制备方法,其中s3步骤中,按重量比计,所述(+)α-(2-噻吩乙胺基)-α-(2-氯苯基)乙酸甲酯盐酸盐与所述甲醛的混合比为1:4~6。
根据本发明所述硫酸氢氯吡格雷晶型ii的制备方法,其中s3步骤中,所述(+)α-(2-噻吩乙胺基)-α-(2-氯苯基)乙酸甲酯盐酸盐与所述甲醛混合后加热,所述加热温度为35~45℃。
根据本发明所述硫酸氢氯吡格雷晶型ii的制备方法,其中s4步骤中,将(+)氯吡格雷游离碱,丙酮和左旋樟脑磺酸混合,搅拌,离心,得(+)氯吡格雷樟脑磺酸复盐。
根据本发明所述硫酸氢氯吡格雷晶型ii的制备方法,其中s5步骤中,将水、无机碱,(+)氯吡格雷樟脑磺酸复盐和二氯甲烷混合,搅拌分液,水层用二氯甲烷萃取,静置分液,合并有机层减压回收二氯甲烷,得(+)氯吡格雷游离碱i。
根据本发明所述硫酸氢氯吡格雷晶型ii的制备方法,其中s6步骤中,加入所述浓硫酸后在0~10℃保温搅拌。
根据本发明所述硫酸氢氯吡格雷晶型ii的制备方法,其中s6步骤中,所述搅拌时间为8~20小时。
根据本发明所述硫酸氢氯吡格雷晶型ii的制备方法,其中s6步骤中,所述搅拌时间为12小时。
根据本发明所述硫酸氢氯吡格雷晶型ii的制备方法,其中s6步骤中,所述无机碱选自磷酸氢二钾、硫酸钠、碳酸钠、碳酸钾、或磷酸氢二钠。
附图说明
图1为使用cu-kα射线测量得到的本发明所述硫酸氢氯吡格雷晶型ii的x-射线粉末衍射图。
图2为使用cu-kα射线测量得到的硫酸氢氯吡格雷晶型ii对照品的x-射线粉末衍射图。
图3为本发明所述硫酸氢氯吡格雷晶型ii的差示热量扫描分析dsc谱图。
图4为硫酸氢氯吡格雷晶型ii对照品的差示热量扫描分析dsc谱图。
图5为本发明所述硫酸氢氯吡格雷晶型ii的tga谱图。
图6为硫酸氢氯吡格雷晶型ii对照品的tga谱图。
具体实施方式
本文中术语“晶型ii”指硫酸氢氯吡格雷化合物在晶体结构中以特定的晶型ii状态存在。不同晶型理化性质的不同可以体现在储存稳定性、可压缩性、密度和溶出速度等方面。溶解度或溶出速度的不同,在极端情况下,可以造成药物低效甚至具有毒性。
不同的晶型可以提供不同的理化特性,比如熔点。熔点的起始温度根据ta分析软件测量得到的dsc基线显著变化的点确定。熔点也可以通过其他技术、其他仪器或其他试验条件来测定。因此,这里熔点的数据并不一定是一个绝对的数值。本领域技术人员会意识到熔点的精确数值会受到化合物的纯度、样品量、加热速度和颗粒度的影响。
本领域技术人员能够理解,这里讨论的理化性质可以通过现有技术手段表征,其中的仪器误差取决于仪器的测试条件、样品的准备和样品纯度。例如,公知x-射线衍射衍射图通常会随着仪器条件的变化而变化,所以峰的强弱顺序通常不予考虑。另外,峰角度的实验误差通常在5%或更少,这些角度的误差通常应该被考虑。因而,可以理解的是,本发明要求保护的晶型的实测x-射线衍射图不必和本文给出的相应x-射线衍射图完全一致。任何具有和本文所给图谱基本相同或相似的图谱的晶型均属于本申请的保护范围。本领域技术人员能够比较本申请所列的图谱和一个未知晶型的图谱,确认这两组图谱反应的是相同还是不同的晶型。
药物的晶型通常可以通过如下方法获得:包括但不限于,熔融重结晶、熔融冷却、溶剂重结晶、脱溶剂、快速挥发、快速降温、慢速降温、蒸气扩散和升华。晶型可以通过x-射线粉末衍射(xrpd)、差示扫描量热分析(dsc)、热重分析(tg)、单晶x射线衍射、振动光谱学、固态nmr、红外光谱、拉曼光谱、溶出速度测定、溶解度、吸湿性等来检测、发现和归类。
本发明检测xrpd谱图所使用的仪器为y-2000x射线衍射仪。样品在室温条件下测试,把需要检测的样品放在有机玻璃片上。检测条件如下,角度范围:30kv;20ma;速度:0.05deg/sec。除非特别说明,样品在检测未经研磨。
本发明检测dsc谱图所使用的仪器为netzschdsc-204型差热分析仪。通常取1-10毫克的样品放置于未加盖(除非特别声明)的铝坩埚内,以10k/min的升温速度在50ml/min干燥n2的保护下将样品从室温升至200℃,记录样品在升温过程中的热量变化。
本发明检测tg谱图所使用的仪器为netzschtg-209。通常取5-15mg的样品放置于白金坩埚内,采用分段高分辨检测的方式,以10k/min的升温速度在50ml/min干燥n2的保护下将样品从室温升至930℃,同时记录样品在升温过程中的热量变化。
本发明提供了一种硫酸氢氯吡格雷晶型ii的制备方法,其包括以下步骤:
s1.制备(+)邻氯苯甘氨酸甲酯
往反应容器里加入萃取剂,水,无机碱,搅拌溶清,再加入(+)邻氯苯甘氨酸甲酯酒石酸盐,反应完全后分液,水层用萃取剂萃取。合并有机层,用干燥剂干燥,离心,取滤液减压回收萃取剂,得(+)邻氯苯甘氨酸甲酯,所述反应过程如下所示:
在本发明的某些具体实施方案中,所述无机碱选自磷酸氢二钾、硫酸钠、碳酸钠、碳酸钾、或磷酸氢二钠。所述无机碱在本发明所述硫酸氢氯吡格雷晶型ii的制备方法中用作缚酸剂,主要是吸收反应步骤中的h+。
在本发明的某些具体实施方案中,所述萃取剂不参与反应,主要用于萃取有机层,可以是能够萃取分层且便于回收的任何与水不互溶的低沸点有机溶剂,包括但不限于,四氢呋喃,二氯甲烷、乙酸乙酯、或苯等,本发明优选二氯甲烷。
s2.制备(+)α-(2-噻吩乙胺基)-α-(2-氯苯基)乙酸甲酯盐酸盐
在本发明的某些具体实施方案中,所述无机碱与所述(+)邻氯苯甘氨酸甲酯的摩尔比为2~2.5:1。
在本发明的某些具体实施方案中,使用盐酸将反应完后离心的母液的ph值调至3~5,在该ph范围内,既能够确保析晶所需要的酸性环境,也能对加酸的量有一个参考,不至于加入大量的酸对环保不利,促使(+)α-(2-噻吩乙胺基)-α-(2-氯苯基)乙酸甲酯进行成盐反应。
同时,在该步骤中,对各温度范围作出准确限定,温度过低,反应不完全,反应的时间要大大延长,有副产物出现的风险;温度过高同样会使得杂质升高。
s3.制备(+)氯吡格雷游离碱
在本发明的某些具体实施方案中,按重量比计,所述(+)α-(2-噻吩乙胺基)-α-(2-氯苯基)乙酸甲酯盐酸盐与所述甲醛的混合比为1:4~6。在本步骤中,甲醛既是溶剂,又是一种反应物。投料太少反应不完全,投料太多造成物料浪费。
在本发明的某些具体实施方案中,所述(+)α-(2-噻吩乙胺基)-α-(2-氯苯基)乙酸甲酯盐酸盐与所述甲醛混合后加热,所述加热温度为35~45℃。
s4.制备(+)氯吡格雷樟脑磺酸复盐
向反应容器里加入(+)氯吡格雷游离碱,丙酮和左旋樟脑磺酸,混合,搅拌,离心,得(+)氯吡格雷樟脑磺酸复盐。
在本发明的某些具体实施方案中,使用左旋樟脑磺酸在丙酮中将所述(+)氯吡格雷游离碱消旋拆分,按重量份计,所述(+)氯吡格雷游离碱:丙酮:左旋樟脑磺酸的混合比为1:4~8:0.5~1。
在上述方案中,丙酮为溶剂;所述左旋樟脑磺酸是反应物之一,若其料量减小,则樟脑磺酸复盐的收率降低,部分(+)氯吡格雷游离碱不能充分成盐而随丙酮母液一起带走。
s5.(+)氯吡格雷樟脑磺酸复盐水解
向反应容器里加入水、无机碱,(+)氯吡格雷樟脑磺酸复盐和二氯甲烷,混合,搅拌分液,水层用二氯甲烷萃取,静置分液,合并有机层减压回收二氯甲烷,得(+)氯吡格雷游离碱i。
在本发明的某些具体实施方案中,按重量份计,所述(+)氯吡格雷樟脑磺酸复盐:二氯甲烷:无机碱=1:2:0.5。在该步骤中,无机碱作为反应物,主要是中和本步骤里面的左旋樟脑磺酸,一般为能接收h+的强碱弱酸盐,其可选自磷酸氢二钾、硫酸钠、碳酸钠、碳酸钾、或磷酸氢二钠。若所述无机碱投料量降低,则氯吡格雷游离碱i的收率降低,部分氯吡格雷樟脑磺酸复盐会随水层一起带走。
尽管s3和s5均为获得氯吡格雷游离碱,但其目的不同。s3中(+)α-(2-噻吩乙胺基)-α-(2-氯苯基)乙酸甲酯盐酸盐与甲醛发生成环反应,然后加入无机碱水解以提高纯度;s5中(+)氯吡格雷樟脑磺酸复盐水解则是消旋拆分,既有纯化的作用也有减少对映异构体的作用,可以将对映异构体含量降至hplc积分面积的0.1%以下。
s6.制备硫酸氢氯吡格雷
在本发明的某些具体实施方案中,按重量比计,所述(+)氯吡格雷游离碱i与丙酮的混合比为1:5~6。
在本发明的某些具体实施方案中,加入浓硫酸后在0~10℃保温搅拌,搅拌时间为8~20小时,优选12小时。
在本发明的某些具体实施方案中,所述干燥温度为75~85℃。
在本发明的某些具体实施方案中,所述干燥时间不低于18小时。时间太短溶剂烘不干,会造成检测溶剂残留超标;时间适当延长,对产品影响不大,因为本产品不含结晶水。
采用本步骤的析晶条件,可以容易地获得高含量的硫酸氢氯吡格雷晶型ii。
与现有技术相比,本发明所述硫酸氢氯吡格雷晶型ii的制备方法,优化硫酸氢氯吡格雷的合成工艺,选用廉价易得的物料,采用温和的反应条件,安全环保的生产出高纯度的产品。本发明所采用的反应原理易于控制,可一次性得到杂质含量较低,对映异构体含量较低的产品。
实施例
为了更好的理解上述技术方案,下面将结合说明书附图和具体的实施例对上述技术方案做详细的说明,但本发明不仅限于这些具体的实施例。
实施例1
向三口烧瓶里加入二氯甲烷300ml,水160ml,碳酸钠80g,搅拌溶清,再加入(+)邻氯苯甘氨酸甲酯酒石酸盐140g,反应完全后分液,水层用50ml二氯甲烷萃取三次,合并有机层,用无水硫酸钠干燥,离心,取滤液减压回收二氯甲烷,得(+)邻氯苯甘氨酸甲酯。
向三口烧瓶里加入600ml乙腈,磷酸氢二钾140g,与上步得到的(+)邻氯苯甘氨酸甲酯混合,20℃搅拌1小时。再加入2-(2-噻吩基)乙醇对甲苯磺酸酯170g,然后升温至48℃,保温反应。反应完后离心,将母液收集至反应容器里,边搅拌边加入适量盐酸至ph为4,搅拌4小时,离心,所得固体50℃干燥,即得(+)α-(2-噻吩乙胺基)-α-(2-氯苯基)乙酸甲酯盐酸盐。
向三口烧瓶里面加入上步得到的(+)α-(2-噻吩乙胺基)-α-(2-氯苯基)乙酸甲酯盐酸盐,4倍重量的甲醛,加热至35℃,保温反应完全。然后冷却降至室温,加入0.5倍重量的硫酸钠、1倍重量的纯化水和6.5倍重量的二氯甲烷,搅拌分液,水层用二氯甲烷萃取两次。合并有机层,减压回收二氯甲烷,得(+)氯吡格雷游离碱。
向三口烧瓶里面加入(+)氯吡格雷游离碱,5倍重量的丙酮和0.65倍重量左旋樟脑磺酸,搅拌24小时,离心,得(+)氯吡格雷樟脑磺酸复盐。
向三口烧瓶里面加入2倍重量的水、0.5倍重量的碳酸钠,(+)氯吡格雷樟脑磺酸复盐和2倍重量二氯甲烷,搅拌分液,水层用二氯甲烷萃取两次,静置分液,合并有机层减压回收二氯甲烷,得(+)氯吡格雷游离碱。
向三口烧瓶里面加入(+)氯吡格雷游离碱,5倍重量的丙酮,过滤,滤液降温加入等摩尔的浓硫酸,0℃保温搅拌12小时,离心,75℃干燥18小时,得到硫酸氢氯吡格雷晶型ii,为白色粉末状固体。
实施例1得到的硫酸氢氯吡格雷晶型ii使用cu-kα射线测量得到的x-射线粉末衍射图,如图1所示,其数据请详见表1。
表1硫酸氢氯吡格雷晶型ii的xrd数据
根据图1的谱图和表1的数据,可以看出,所述x射线粉末衍射2θº衍射角位置在21.7处峰强比为100,其次在23.0和24.8处有强峰。
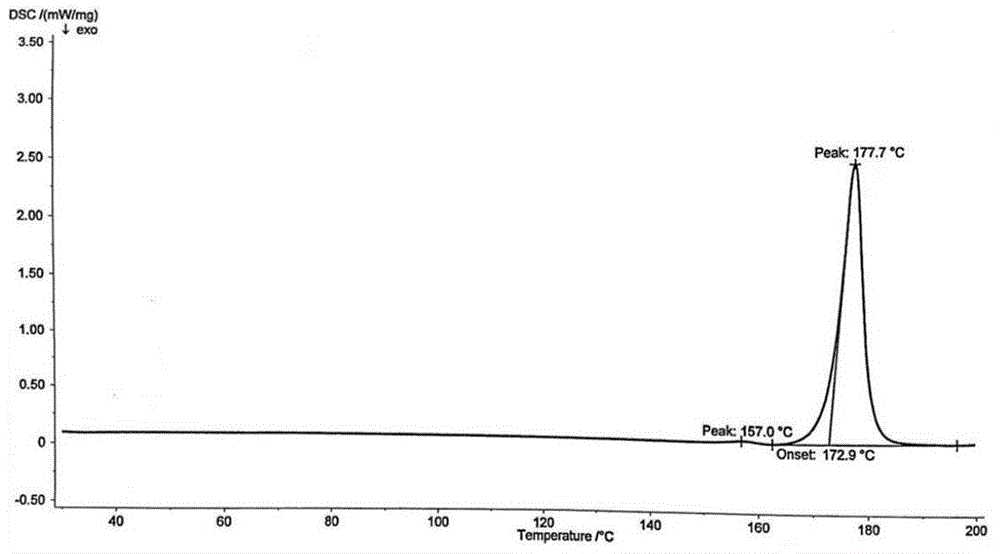
对该样品进行差热分析,dsc结果表明,该样品在177.5℃处有明确的相变点,结果如图3所示。
对该样品进行tg分析,tg结果表明,该样品在100℃时几乎无失重,在192.3℃仅失重1%,表明被检样品中不含有吸附水,结果如图5所示。
对比例1
图2中的对照品由中国食品药品检定研究院提供。
对比例1得到的对比样品使用cu-kα射线测量得到的x-射线粉末衍射图,如图2所示,其数据请详见表2。
表2硫酸氢氯吡格雷晶型ii的xrd数据
根据图2的谱图和表2的数据,可以看出,所述x射线粉末衍射2θº衍射角位置在21.5处峰强比为100,其次在22.9和24.6处有强峰。
对该样品进行差热分析,dsc结果表明,与实施例1样品相同,该样品也在177.5℃处有明确的相变点,结果如图4所示。
对该样品进行tg分析,tg结果表明,与实施例1样品相同,该样品在100℃时几乎无失重,在192.3℃仅失重1%,表明被检样品中不含有吸附水,结果如图6所示。
由此可见,根据本发明所述制备方法获得的实施例1样品与对比例1中的样品的谱图和数据基本一致。
实施例2
向三口烧瓶里加入二氯甲烷300ml,水160ml,碳酸钠80g,搅拌溶清,再加入(+)邻氯苯甘氨酸甲酯酒石酸盐140g,反应完全后分液,水层用50ml二氯甲烷萃取三次,合并有机层,用无水硫酸钠干燥,离心,取滤液减压回收二氯甲烷,得(+)邻氯苯甘氨酸甲酯。
向三口烧瓶里加入600ml乙腈,磷酸氢二钾140g,与上步得到的(+)邻氯苯甘氨酸甲酯混合,30℃搅拌1小时。再加入2-(2-噻吩基)乙醇对甲苯磺酸酯170g,然后升温至52℃,保温反应。反应完后离心,将母液收集至反应容器里,边搅拌边加入适量盐酸至ph为4,搅拌4小时,离心,所得固体60℃干燥,即得(+)α-(2-噻吩乙胺基)-α-(2-氯苯基)乙酸甲酯盐酸盐。
向三口烧瓶里面加入上步得到的(+)α-(2-噻吩乙胺基)-α-(2-氯苯基)乙酸甲酯盐酸盐,6倍重量的甲醛,加热至45℃,保温反应完全。然后冷却降至室温,加入0.5倍重量的硫酸钠、1倍重量的纯化水和6.5倍重量的二氯甲烷,搅拌分液,水层用二氯甲烷萃取两次。合并有机层,减压回收二氯甲烷,得(+)氯吡格雷游离碱。
向三口烧瓶里面加入(+)氯吡格雷游离碱,5倍重量的丙酮和0.65倍重量左旋樟脑磺酸,搅拌24小时,离心,得(+)氯吡格雷樟脑磺酸复盐。
向三口烧瓶里面加入2倍重量的水、0.5倍重量的碳酸钠,(+)氯吡格雷樟脑磺酸复盐和2倍重量二氯甲烷,搅拌分液,水层用二氯甲烷萃取两次,静置分液,合并有机层减压回收二氯甲烷,得(+)氯吡格雷游离碱。
向三口烧瓶里面加入(+)氯吡格雷游离碱,5倍重量的丙酮,过滤,滤液降温加入等摩尔的浓硫酸,10℃保温搅拌12小时,离心,85℃干燥20小时,得到硫酸氢氯吡格雷晶型ii,为白色粉末状固体。
对比例2
向三口烧瓶里加入二氯甲烷300ml,水160ml,碳酸钠80g,搅拌溶清,再加入(+)邻氯苯甘氨酸甲酯酒石酸盐140g,反应完全后分液,水层用50ml二氯甲烷萃取三次,合并有机层,用无水硫酸钠干燥,离心,取滤液减压回收二氯甲烷,得(+)邻氯苯甘氨酸甲酯。
向三口烧瓶里加入600ml乙腈,磷酸氢二钾140g,与上步得到的(+)邻氯苯甘氨酸甲酯混合,30℃搅拌1小时。再加入2-(2-噻吩基)乙醇对甲苯磺酸酯170g,然后升温至52℃,保温反应。反应完后离心,将母液收集至反应容器里,边搅拌边加入适量盐酸至ph为4,搅拌4小时,离心,所得固体60℃干燥,即得(+)α-(2-噻吩乙胺基)-α-(2-氯苯基)乙酸甲酯盐酸盐。
向三口烧瓶里面加入上步得到的(+)α-(2-噻吩乙胺基)-α-(2-氯苯基)乙酸甲酯盐酸盐,6倍重量的甲醛,加热至45℃,保温反应完全。然后冷却降至室温,加入0.5倍重量的硫酸钠、1倍重量的纯化水和6.5倍重量的二氯甲烷,搅拌分液,水层用二氯甲烷萃取两次。合并有机层,减压回收二氯甲烷,得(+)氯吡格雷游离碱。
向三口烧瓶里面加入(+)氯吡格雷游离碱,5倍重量的丙酮和0.65倍重量左旋樟脑磺酸,搅拌24小时,离心,得(+)氯吡格雷樟脑磺酸复盐。
向三口烧瓶里面加入2倍重量的水、0.5倍重量的碳酸钠,(+)氯吡格雷樟脑磺酸复盐和2倍重量二氯甲烷,搅拌分液,水层用二氯甲烷萃取两次,静置分液,合并有机层减压回收二氯甲烷,得(+)氯吡格雷游离碱。
向三口烧瓶里面加入(+)氯吡格雷游离碱,5倍重量的丙酮,过滤,滤液降温加入等摩尔的浓硫酸,20℃保温搅拌12小时,离心,85℃干燥20小时,得到硫酸氢氯吡格雷晶型ii,为棕褐色粉末状固体。
稳定性实验
对实施例1制备得到的硫酸氢氯吡格雷晶型ii进行稳定性考察,其结果如表3所示:
表3硫酸氢氯吡格雷原料药的长期稳定性研究数据
其中,杂质i是杂质a;杂质ii是杂质;单杂是指除已知杂质外的最大未知杂质。
尽管已描述了本发明的优选实施例,但本领域内的技术人员一旦得知了基本创造性概念,则可对这些实施例作出另外的变更和修改。所以,所附权利要求意欲解释为包括优选实施例以及落入本发明范围的所有变更和修改。显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。
- 还没有人留言评论。精彩留言会获得点赞!