一种稳态化花翠素-3-O-葡萄糖苷衍生物及制备方法与流程
本发明属于化合物及其制备技术领域,具体涉及一种稳态化花翠素-3-o-葡萄糖苷衍生物及制备方法。
背景技术:
花青素,又称花色苷,广泛存在于植物中,水果、蔬菜、花卉、谷物中都含有丰富的花青素,如蓝莓、葡萄、蓝靛果、越橘、桑葚、草莓、紫甘薯、紫玉米、紫甘蓝、黑豆、黑米、玫瑰茄等。花青素在葡萄、蓝莓、蓝靛果中含量较高,尤其是山葡萄,可作为天然花青素的很好来源。花青素属类黄酮化合物,其中常见花青素类别包括天竺葵素、矢车菊素、飞燕草素、芍药素、矮牵牛素、锦葵素等。花青素作为一种强有力的天然抗氧化剂,除与黄酮类化合物清除自由基的机制一致外,花青素类还有自己的特殊性质,其在酸性条件下带正电,天然缺电子的特性使其反应活性很高,使其在清除自由基方面更有优势;对于人类慢性疾病治疗如糖尿病、高血压等都具有广阔的空间,同时对于降低心血管疾病的危险率、肥胖症的发生率都具有积极地作用,为人类的健康带来了极大的帮助。
天然的花青素色泽艳丽,不仅在食品加工等领域应用潜力大,而且广泛应用于功能性食品及保健食品领域中。但天然花青素稳定性差,易受温度、光照、ph、氧、酶、金属离子等影响,在提取、加工及贮藏过程中易发生降解,出现褪色、变色、沉淀等现象,使其应用受限。现有技术中,提高花青素稳定性的方法包括辅色、微胶囊化、生物工程技术等,但因受技术、条件、政策等多方面限制,这些方法仍无法满足生产需要;如有机酸与花青素之间的互作辅色技术是提高花青素稳定性的重要途径,但自发性辅色反应的速度慢,效率低,无法满足实际生产的需要。
超高压是把液体或气体加压到100mpa以上的非热加工技术,主要用于杀菌、成分提取、催陈等。现有技术中,关于超高压技术应用于辅色花青素增加其稳定性的研究甚少,且超高压促花翠素-3-o-葡萄糖苷与咖啡酸稳态化处理及其机理未见报道。
技术实现要素:
有鉴于此,本发明为解决现有技术中花青素稳定性差的技术问题,提供一种稳态化花翠素-3-o-葡萄糖苷衍生物及制备方法。
本发明解决上述技术问题采取的技术方案如下。
本发明提供一种稳态化花翠素-3-o-葡萄糖苷衍生物,结构式如式ⅰ所示:
本发明还提供上述稳态化花翠素-3-o-葡萄糖苷衍生物的制备方法,包括如下步骤:
步骤一、将山葡萄去梗,打浆,得山葡萄浆液;
步骤二、采用非热提取技术提取山葡萄浆液中的花青素,得花青素溶液;
所述非热提取技术为超声提取或高压脉冲电场提取;
步骤三、将花青素溶液离心,取上清液,经减压浓缩,大孔树脂洗脱,合并洗脱液,减压浓缩,冷冻干燥,得花青素纯化物;
步骤四、用含0.5~1%(v/v)hcl的甲醇溶液溶解花青素纯化物,使用制备液相色谱分离,收集洗脱峰,冷冻干燥,得花翠素-3-o-葡萄糖苷;
步骤五、将咖啡酸和花翠素-3-o-葡萄糖苷按照质量比为1:(3~5)溶于有机溶剂中,混合均匀,得到混合液,使用ph为3.0~4.0的缓冲液稀释混合液至花翠素-3-o-葡萄糖苷的浓度为0.1~0.2mg/ml;先在避光下,50~60℃水浴30~60min,然后采用超高压稳态化处理,稳定压力160~500mpa,保压时间1~15min,得到的处理液经大孔树脂柱纯化后,减压浓缩,冷冻干燥,得稳态化花翠素-3-o-葡萄糖苷衍生物。
优选的是,所述步骤二中,超声提取工艺为:提取溶剂为0.1%盐酸-75%乙醇溶液,液料比为7:1w/w,功率为270w,时间为15min。
优选的是,所述步骤二中,高压脉冲电场提取工艺为:提取溶剂为0.1%盐酸-75%乙醇溶液,液料比为9:1w/w,电场强度为15kv/cm,脉冲数为4。
优选的是,所述步骤三中,大孔树脂的型号为d101,大孔树脂洗脱的过程为:上样及洗脱速率为1.5bv/h,上样后静置30min,用5~8倍柱体积的纯化水洗脱杂质,再用0.1%hcl-75%乙醇溶液洗脱至无色。
优选的是,所述步骤三中,取上清液减压浓缩至相对密度为1.1~1.2,温度为50℃;合并洗脱液减压浓缩至相对密度为1.2~1.3,温度为50℃。
优选的是,所述步骤四中,制备液相色谱分离的条件为:
色谱柱:sunfireprepc18柱,尺寸为19mm×50mm,5μm;检测波长530nm;柱温25℃;流速5ml/min;进样量150μl;流动相a-5%甲酸水溶液,b-甲醇,梯度洗脱:0~5min,15%b;5~8min,15%~20%b;8~13min,10%~15%b;13~16min,25%~31%b;16~17min,31%~100%b;17~19min,100%b;19~20min,100%~15%b;20~23min,15%b。
优选的是,所述步骤五中,花翠素-3-o-葡萄糖苷的浓度为0.1mg/ml。
优选的是,所述步骤五中,水浴温度为55℃,水浴时间为45min。
优选的是,所述步骤五中,稳定压力为200~400mpa。
优选的是,所述步骤五中,减压浓缩至相对密度为1.2~1.3,温度为50℃。
优选的是,所述步骤五中,有机溶剂为无水乙醇。
优选的是,所述步骤五中,缓冲液为磷酸氢二钠-柠檬酸缓冲液或醋酸-醋酸钠缓冲液。
与现有技术相比,本发明的有益效果为:
本发明的稳态化花翠素-3-o-葡萄糖苷衍生物的制备方法采用超声提取或高压脉冲电场提取山葡萄中的花青素,最大程度保护花青素不受破坏;制备的产物为花翠素-3-o-(6″-o-咖啡酰基)-葡萄糖苷,采用超高压稳态化处理,相比花翠素-3-o-葡萄糖苷,具有更好的光热稳定性,且具备抗氧化作用和抗肿瘤作用。经实验检测,花翠素-3-o-(6″-o-咖啡酰基)-葡萄糖苷的增色率可达40%以上,在100℃加热2h及在室温日光放置20d后,其保存率r值均可达60%以上;不同浓度的花翠素-3-o-(6″-o-咖啡酰基)-葡萄糖苷对氧化损伤huvec细胞处理后,细胞增殖率显著增加,活性氧显著降低;对hepg-2肝癌细胞、hl-60急性早幼粒白血病细胞处理后,细胞抑制率显著增加。
附图说明
为了更清楚地说明本发明实施例中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其它的附图。
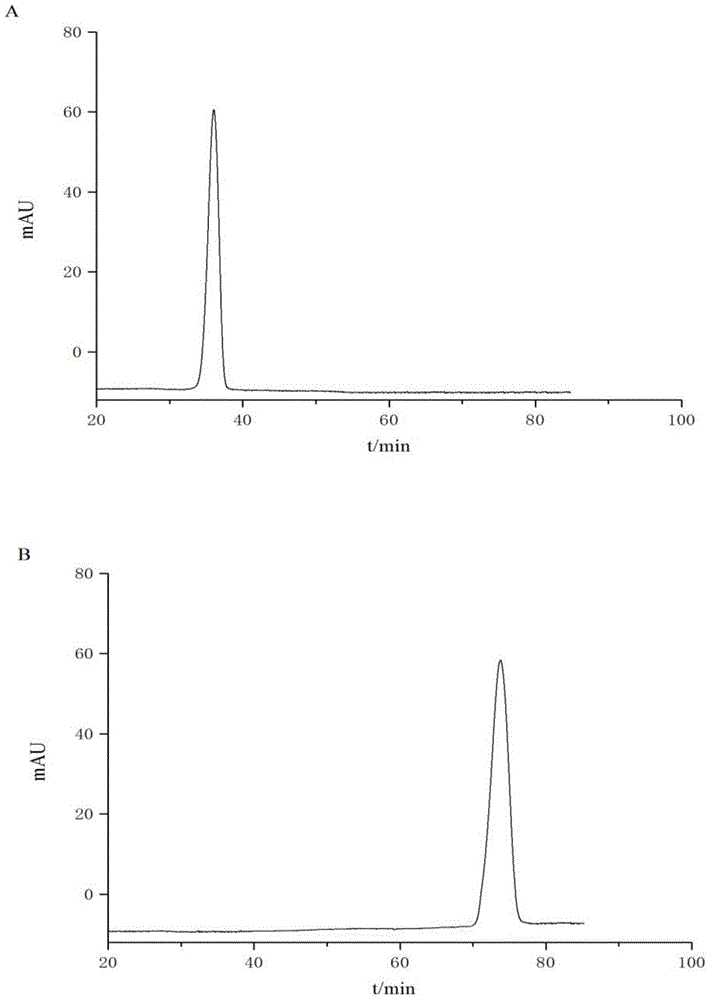
图1中,a和b分别为实施例1的花翠素3-o-葡萄糖苷及花翠素-3-o-葡萄糖苷衍生物的hplc图谱,c为对比例1制备的产物的hplc图谱;
图2为实施例1的花翠素-3-o-葡萄糖苷衍生物的ms/ms图谱。
具体实施方式
为了进一步了解本发明,下面结合具体实施方式对本发明的优选实施方案进行描述,但是应当理解,这些描述只是为进一步说明本发明的特征和优点而不是对本发明专利要求的限制。
本发明的稳态化花翠素-3-o-葡萄糖苷衍生物,结构式如式ⅰ所示:
本发明的稳态化花翠素-3-o-葡萄糖苷衍生物的制备方法,包括如下步骤:
步骤一、将山葡萄去梗,打浆,得山葡萄浆液;
步骤二、采用非热提取技术提取山葡萄浆液中的花青素,得花青素溶液;
步骤三、将花青素溶液离心,取上清液,经减压浓缩,大孔树脂洗脱,合并洗脱液,减压浓缩,冷冻干燥,得花青素纯化物;
步骤四、用含0.5~1%(v/v)hcl的甲醇溶液溶解花青素纯化物,使用制备液相色谱分离,收集洗脱峰,冷冻干燥,得花翠素-3-o-葡萄糖苷;
步骤五、将咖啡酸和花翠素-3-o-葡萄糖苷按照质量比为1:(3~5)溶于有机溶剂中,混合均匀,得到混合液,使用ph为3.0~4.0的缓冲液稀释混合液至花翠素-3-o-葡萄糖苷的浓度为0.1~0.2mg/ml;先在避光下,50~60℃水浴30~60min,然后采用超高压稳态化处理,稳定压力200~400mpa,保压时间1~15min,得到的处理液经大孔树脂柱纯化后,减压浓缩,冷冻干燥,得稳态化花翠素-3-o-葡萄糖苷衍生物。
本发明的原理为:稳态化3-o-葡萄糖苷衍生物的形成机理为:花翠素-3-o-葡萄糖苷与咖啡酸键合,脱去一分子h2o得花翠素-3-o-(6″-o-咖啡酰基)-葡萄糖苷,反应过程如下:
上述技术方案,步骤一中,山葡萄没有特殊限制,本领域技术人员能够依据熟知方式获得。花青素含量越高的山葡萄越好,优选采用长白山野生山葡萄。山葡萄去梗和打浆的具体操作为本领域技术人员熟知技术,工艺没有特殊限制。
上述技术方案,步骤二中,非热提取技术为超声提取或高压脉冲电场提取;超声提取工艺和高压脉冲电场提取为本领域技术人员熟知的非热提取技术,本发明研究,当采用以下工艺时,效果最好,故优选超声提取工艺为:提取溶剂0.1%盐酸-75%乙醇溶液,液料比7:1w/w,功率270w,时间15min;优选高压脉冲电场提取工艺为:提取溶剂0.1%盐酸-75%乙醇溶液,液料比9:1w/w,电场强度15kv/cm,脉冲数4。
上述技术方案,步骤三中,离心、减压浓缩和冷冻干燥的条件没有特殊限制。优选离心条件为:4500r/min离心10min;离心后取上清液的方法可以为缓慢倾倒出上清液,也可以采用过滤的方法分离出上清液。优选取上清液减压浓缩至相对密度为1.1~1.2,温度为50℃。优选合并洗脱液减压浓缩至相对密度为1.2~1.3,温度为50℃。优选冷冻干燥的条件为:预冻结温度-35℃,加热温度40℃,真空度30pa。大孔树脂洗脱采用的大孔树脂的型号优选为d101。大孔树脂洗脱,合并洗脱液的工艺为:上样及洗脱速率为1.5bv/h,上样后静置30min,用5~8倍柱体积纯化水洗脱杂质,再用0.1%hcl-75%乙醇洗脱至无色,合并洗脱液。
上述技术方案,步骤四中,使用制备液相色谱分离花翠素-3-o-葡萄糖苷为现有技术,本领域技术人员可以根据花翠素-3-o-葡萄糖苷的保留时间将其分离。本发明提供一种制备液相色谱分离的条件,但不限于此:色谱柱:sunfireprepc18柱,尺寸为19mm×50mm,5μm;检测波长530nm;柱温25℃;流速5ml/min;进样量150μl;流动相a-5%甲酸水溶液,b-甲醇,梯度洗脱:0~5min,15%b;5~8min,15%~20%b;8~13min,10%~15%b;13~16min,25%~31%b;16~17min,31%~100%b;17~19min,100%b;19~20min,100%~15%b;20~23min,15%b。冷冻干燥的条件没有特殊限制,优选预冻结温度-35℃,加热温度40℃,真空度30pa。
上述技术方案,步骤五中,咖啡酸和花翠素-3-o-葡萄糖苷质量比为1:(3-5),如果花翠素-3-o-葡萄糖苷加入量少于咖啡酸质量的3倍,产物中会含有未键合的花翠素-3-o-葡萄糖苷,如果花翠素-3-o-葡萄糖苷的加入量大于咖啡酸质量的5倍,产物中也会含有未键合的花翠素-3-o-葡萄糖苷。有机溶剂优选为无水乙醇,溶解顺序没有特殊限制,一般可先溶解咖啡酸,再加入花翠素-3-o-葡萄糖苷。混合均匀的方式没有特殊限制,可采用机械搅拌混合。水浴温度为50~60℃,如果水浴温度低于50℃,制备的衍生物的增色率及光、热稳定性均降低;花青素本身容易降解,温度越高,降解速度越快,如果水浴温度高于60℃,容易造成花青素降解;优选水浴温度为55℃,水浴时间为45min。超高压稳态化处理的稳定压力为160-500mpa,制备的衍生物的增色率及光、热稳定性随稳定压力的升高先升高后降低,如果稳定压力小于160mpa或大于500mpa,制备的衍生物的增色率及光、热稳定性均不理想,当稳定压力为200-400mpa时,制备的衍生物的增色率及光、热稳定性效果较优,故优选稳定压力为200-400mpa,当稳态压力为300mpa,制备的衍生物的增色率及光、热稳定性效果最优,故最优选稳定压力为300mpa。保压时间优选为3~7min,更优选为5min。优选花翠素-3-o-葡萄糖苷的浓度为0.1mg/ml。减压浓缩和冷冻干燥的条件没有特殊限制。优选减压浓缩至相对密度为1.2~1.3,温度为50℃。优选预冻结温度-35℃,加热温度40℃,真空度30pa。缓冲液优选为磷酸氢二钠-柠檬酸缓冲液或醋酸-醋酸钠缓冲液。
以下通过实施例对本发明作进一步的阐述。
实施例1
步骤一、将山葡萄去梗,打浆,得山葡萄浆液;
步骤二、采用高压脉冲电场提取山葡萄浆液中的花青素,得花青素溶液;其中,提取溶剂为0.1%盐酸-75%乙醇溶液,液料比9:1w/w,电场强度15kv/cm,脉冲数4;
步骤三、将花青素溶液离心,取上清液,经减压浓缩(至相对密度为1.1,50℃),大孔树脂洗脱,合并洗脱液,减压浓缩(至相对密度为1.2,50℃),冷冻干燥,得花青素纯化物;其中,制备液相色谱分离的条件为:色谱柱:sunfireprepc18柱,尺寸为19mm×50mm,5μm;检测波长530nm;柱温25℃;流速5ml/min;进样量150μl;流动相a-5%甲酸水溶液,b-甲醇,梯度洗脱:0~5min,15%b;5~8min,15%~20%b;8~13min,10%~15%b;13~16min,25%~31%b;16~17min,31%~100%b;17~19min,100%b;19~20min,100%~15%b;20~23min,15%b;
步骤四、用含0.5~1%(v/v)hcl的甲醇溶液溶解花青素纯化物,使用制备液相色谱分离,收集洗脱峰,冷冻干燥,得花翠素-3-o-葡萄糖苷;
步骤五、用无水乙醇溶解咖啡酸,加入质量为咖啡酸质量3倍的花翠素-3-o-葡萄糖苷,混合均匀,制得混合液;用ph为3.0的磷酸氢二钠-柠檬酸缓冲液稀释上述混合液,使花翠素-3-o-葡萄糖苷的浓度为0.1mg/ml,避光下,50℃水浴30min后,进行超高压稳态化处理,稳定压力300mpa,保压时间5min,所得处理液经大孔树脂柱纯化后,减压浓缩(至相对密度为1.2,50℃),冷冻干燥,得稳态化花翠素-3-o-葡萄糖苷衍生物。
实施例2
步骤一、将山葡萄去梗,打浆,得山葡萄浆液;
步骤二、采用高压脉冲电场提取山葡萄浆液中的花青素,得花青素溶液;其中,提取溶剂为0.1%盐酸-75%乙醇溶液,液料比9:1w/w,电场强度15kv/cm,脉冲数4;
步骤三、将花青素溶液离心,取上清液,经减压浓缩(至相对密度为1.2,50℃),大孔树脂洗脱,合并洗脱液,减压浓缩(至相对密度为1.3,50℃),冷冻干燥,得花青素纯化物;
步骤四、用含0.5~1%(v/v)hcl的甲醇溶液溶解花青素纯化物,使用制备液相色谱分离,收集洗脱峰,冷冻干燥,得花翠素-3-o-葡萄糖苷;其中,制备液相色谱分离的条件为:色谱柱:sunfireprepc18柱,尺寸为19mm×50mm,5μm;检测波长530nm;柱温25℃;流速5ml/min;进样量150μl;流动相a-5%甲酸水溶液,b-甲醇,梯度洗脱:0~5min,15%b;5~8min,15%~20%b;8~13min,10%~15%b;13~16min,25%~31%b;16~17min,31%~100%b;17~19min,100%b;19~20min,100%~15%b;20~23min,15%b;
步骤五、用无水乙醇溶解咖啡酸,加入质量为咖啡酸质量的4倍的花翠素-3-o-葡萄糖苷,混合均匀,制得混合液;用ph为3.0的磷酸氢二钠-柠檬酸缓冲液稀释上述混合液,使花翠素-3-o-葡萄糖苷的浓度为0.1mg/ml,避光下,50℃水浴30min;进行超高压稳态化处理,稳定压力300mpa,保压时间5min,所得处理液经大孔树脂柱纯化后,减压浓缩(至相对密度为1.3,50℃),冷冻干燥,得稳态化花翠素-3-o-葡萄糖苷衍生物。
实施例3
步骤一、将山葡萄去梗,打浆,得山葡萄浆液;
步骤二、采用高压脉冲电场提取山葡萄浆液中的花青素,得花青素溶液;其中,提取溶剂为0.1%盐酸-75%乙醇溶液,液料比9:1w/w,电场强度15kv/cm,脉冲数4;
步骤三、将花青素溶液离心,取上清液,经减压浓缩(至相对密度为1.2,50℃),大孔树脂洗脱,合并洗脱液,减压浓缩(至相对密度为1.3,50℃),冷冻干燥,得花青素纯化物;
步骤四、用含0.5~1%(v/v)hcl的甲醇溶液溶解花青素纯化物,使用制备液相色谱分离,收集洗脱峰,冷冻干燥,得花翠素-3-o-葡萄糖苷;其中,制备液相色谱分离的条件为:色谱柱:sunfireprepc18柱,尺寸为19mm×50mm,5μm;检测波长530nm;柱温25℃;流速5ml/min;进样量150μl;流动相a-5%甲酸水溶液,b-甲醇,梯度洗脱:0~5min,15%b;5~8min,15%~20%b;8~13min,10%~15%b;13~16min,25%~31%b;16~17min,31%~100%b;17~19min,100%b;19~20min,100%~15%b;20~23min,15%b;
步骤五、用无水乙醇溶解咖啡酸,加入质量为咖啡酸质量的5倍的花翠素-3-o-葡萄糖苷,混合均匀,制得混合液;用ph为3.6的醋酸-醋酸钠缓冲液稀释上述混合液,使花翠素-3-o-葡萄糖苷的浓度为0.1mg/ml,避光下,50℃水浴30min;进行超高压稳态化处理,稳定压力300mpa,保压时间5min,所得处理液经大孔树脂柱纯化后,减压浓缩(至相对密度为1.2,50℃),冷冻干燥,得稳态化花翠素-3-o-葡萄糖苷衍生物。
实施例4
步骤一、将山葡萄去梗,打浆,得山葡萄浆液;
步骤二、采用超声提取山葡萄浆液中的花青素,得花青素溶液;其中,提取溶剂0.1%盐酸-75%乙醇溶液,液料比7:1w/w,功率270w,时间15min;
步骤三、将花青素溶液离心,取上清液,经减压浓缩(至相对密度为1.2,50℃),大孔树脂洗脱,合并洗脱液,减压浓缩(至相对密度为1.3,50℃),冷冻干燥,得花青素纯化物;
步骤四、用含0.5~1%(v/v)hcl的甲醇溶液溶解花青素纯化物,使用制备液相色谱分离,收集洗脱峰,冷冻干燥,得花翠素-3-o-葡萄糖苷;其中,制备液相色谱分离的条件为:色谱柱:sunfireprepc18柱,尺寸为19mm×50mm,5μm;检测波长530nm;柱温25℃;流速5ml/min;进样量150μl;流动相a-5%甲酸水溶液,b-甲醇,梯度洗脱:0~5min,15%b;5~8min,15%~20%b;8~13min,10%~15%b;13~16min,25%~31%b;16~17min,31%~100%b;17~19min,100%b;19~20min,100%~15%b;20~23min,15%b;
步骤五、用无水乙醇溶解咖啡酸,加入质量为咖啡酸质量的3倍的花翠素-3-o-葡萄糖苷,混合均匀,制得混合液;用ph为4.0的醋酸-醋酸钠缓冲液稀释上述混合液,使花翠素-3-o-葡萄糖苷的浓度为0.2mg/ml,避光下,60℃水浴60min;进行超高压稳态化处理,稳定压力300mpa,保压时间5min,所得处理液经大孔树脂柱纯化后,减压浓缩(至相对密度为1.2,50℃),冷冻干燥,得稳态化花翠素-3-o-葡萄糖苷衍生物。
实施例5
将实施例3步骤五中,稳定压力替换为250mpa,其他条件同实施例3。
实施例6
将实施例3步骤五中,稳定压力替换为350mpa,其他条件同实施例3。
实施例7
将实施例3步骤五中,保压时间替换为3min,其他条件同实施例3。
实施例8
将实施例3步骤五中,保压时间替换为7min,其他条件同实施例3。
对比例1
将实施例1步骤五中,花翠素-3-o-葡萄糖苷加入量替换为质量为咖啡酸质量的1倍,其他条件同实施例1。
对比例2
将实施例1步骤五中,花翠素-3-o-葡萄糖苷加入量替换为质量为咖啡酸质量的2倍,其他条件同实施例1。
对比例3
将实施例1步骤五中,花翠素-3-o-葡萄糖苷加入量替换为质量为咖啡酸质量的6倍,其他条件同实施例1。
对比例4
将实施例1步骤五中,稳定压力替换为150mpa,其他条件同实施例1。
对比例5
将实施例1步骤五中,稳定压力替换为550mpa,其他条件同实施例1。
对比例6
将实施例1步骤五中,水浴温度替换为30℃,其他条件同实施例1。
1.1对实施例1~8和对比例1~6制备的衍生物进行结构鉴定。
分析色谱条件为色谱柱:thermosyncronisc18色谱柱(100mm×3mm,1.7μm),梯度洗脱,流动相:0.1%甲酸水溶液(a)和乙腈(b);梯度洗脱:0~5min,8~10%b;5~23min,10%b;23~38min,10~15%b;38~48min,15~20%b;48~53min,20~25%b;53~66min,25~35%b;66~71min,35~45%b;71~76min,45~55%b;76~80min,55~80%b;柱温30℃;流速0.2ml·min-1;进样量5μl。质谱条件为采用电喷雾正离子扫描模式(esi+),干燥气温度350℃,雾化气流速35arb,辅助气流速10arb,质量扫描范围m/z50~1000。结果如图1和图2所示。
图1中,a和b分别为实施例1的花翠素3-o-葡萄糖苷及花翠素-3-o-葡萄糖苷衍生物的hplc图谱;图2为实施例1中花翠素-3-o-葡萄糖苷衍生物的ms/ms图谱。从图1和图2可以看出,在正离子模式下,花翠素-3-o-葡萄糖苷与咖啡酸键合衍生物存在准分子离子峰m/z627.13562,在二级串联质谱中可见碎片离子m/z465.13562,303.13525,141.13497。其中,碎片离子m/z465.13562是由分子离子m/z627.13562失去1分子质量数为162的咖啡酰基产生的,碎片离子m/z303.13525是由碎片离子m/z465.13562继续丢失1分子质量数为162的葡萄糖残基产生的;m/z141.13497是由花青素c环发生0/2位c-c键断裂而产生的0,2a+·自由基正离子。根据相关文献报道并进行综合分析,鉴定为花翠素-3-o-(6"-o-咖啡酰基)-葡萄糖苷。实施例2~8的分析结果与实施例1类似。通过hplc-ms/ms联用技术对对比例1~6制得的产物进行结构鉴定,图1中,c为对比例1制备的产物的hplc图谱,从图1可以看出,对比例1制备的产物中共检测出两种化合物,与实施例1相比较,均含有未键合的花翠素-3-o-葡萄糖苷。对比例2~6制备的产物的分析结果与对比例1类似。
1.2对实施例1~8和对比例1~6制备的衍生物进行稳定性评价。
以未高压键合处理的花翠素作为对照,在λ521nm处测定吸光度值,按公式1-1计算稳态化后的增色率i(%),评估其反应效果。
i(%)=[(a-a0)/a0]×100公式(1-1)
式中:a为稳态化花翠素-3-o-葡萄糖苷溶液的吸光度值;a0为未处理花翠素-3-o-葡萄糖苷溶液的吸光度值。
以未高压键合处理的花翠素作为对照,样品分别在100℃条件下处理2.0h、日光条件下处理20d,分别在λ521nm处测定吸光度值,按公式1-2计算稳态化后的保存率r(%)。
r(%)=(at/a)×100公式(1-2)
式中:at为光、热不同处理时间后稳态化花翠素-3-o-葡萄糖苷溶液的吸光度值;a为光、热处理前稳态化花翠素-3-o-葡萄糖苷溶液的吸光度值。
结果如表1和表2所示。从表1可以看出,实施例1~3制备的衍生物的增色率可达40%以上;在100℃加热2h及在室温日光放置20d后,保存率r值均可达60%以上。从表2可以看出,与实施例1~8制备的衍生物相比较,对比例1~6制备的衍生物的增色率及光稳定性均降低。
表1实施例1~8制备的衍生物的稳定性评价结果
表2对比例1~6制备的衍生物的稳定性评价结果
1.3对实施例1~3制备的衍生物进行抗氧化活性试验。
试验采用细胞试验,人脐静脉血管内皮细胞(huvec)的培养:将huvec细胞从液氮中取出,快速放入37℃水浴锅中,轻摇冻存管使冻存液溶解;细胞转移到含有5ml培养基的离心管中,室温1000rpm/min离心5min,弃上清;用含10%胎牛血清的细胞培养基悬浮细胞,接种到培养皿中,轻轻吹打,混匀,细胞置于37℃、5%co2饱和湿度条件下培养。当细胞的密度达到80%时,对细胞进行传代。弃去培养基,用pbs缓冲液洗一遍;加1~2ml0.25%胰蛋白酶消化细胞,显微镜下观察,消化1~2min,可看到细胞相互分离变圆,即消化完成;最后,快速弃去胰酶,加入完全培养基,吹打细胞,制成单细胞悬液,按1:3的比例传代,细胞置于37℃、5%co2饱和湿度条件下扩大培养。选择生长状态良好4~9代细胞进行试验。
cck-8检测细胞增殖率:取处于对数生长期,生长状态良好的huvec细胞,按5×103个/孔接种于96孔板中,每孔加入培养液200μl,三个平行样,置于37℃、5%co2培养箱中培养24h;空白组、模型组、对照组、实施例组均处理24h,每孔加入20μlcck-8溶液,37℃培养4h,酶标仪测定各孔吸光值od450。
细胞增殖率(%)=(od样品组/od对照组)×100%公式(1-3)
细胞抑制率(%)=1-(od样品组/od对照组)×100%公式(1-4)
细胞内ros水平的测定:取处于对数生长期,生长状态良好的huvec细胞,按5×103个/孔接种于96孔板中,每孔加入培养液200μl,37℃、5%co2培养箱中培养24h,空白组、模型组、对照组、实施例组均处理24h,用pbs缓冲液将细胞润洗1次;然后按照dcfh-da细胞ros检测试剂盒操作说明进行,用无血清培养液按1:1000稀释dcfh-da,使终浓度为10μmol,细胞加入稀释好的dcfh200μl,37℃培养箱孵育20min,每隔3min混匀一次,再用无血清培养基洗涤细胞2次,pbs缓冲液重悬,酶标仪荧光检测(激发波长为480nm,发射波长为525nm)。
比值(ratio,r)=a样品组/a对照组公式(1-5)
式中:a样品组为受试组的吸光度值;a对照组为对照组的吸光度值。
huve细胞氧化损伤模型的建立:分别考察h2o2浓度为100μmol/l、200μmol/l、300μmol/l、500μmol/l、750μmol/l、1000μmol/l对huvec细胞进行不同时间(6h、12h、24h、48h)作用处理后,采用cck-8法检测细胞抑制率。每组设3个复孔,结果取平均值,计算不同浓度h2o2对细胞增殖的抑制率(%),以细胞增殖抑制率为50%(ic50)的h2o2浓度作为后续实验的h2o2浓度,并检测细胞内ros的水平。结果如表3所示,随着处理时间的延长,h2o2诱导huvec细胞氧化应激损伤的半数抑制浓度ic50逐渐降低,呈现出浓度依赖性;不同浓度h2o2对huvec细胞进行不同时间处理后,细胞内ros水平测定结果如表4所示,而当h2o2溶液浓度为300μmol/l时,细胞中的ros比值与空白组比较开始有显著性差异(p<0.05),培养24h及48h时,细胞中的ros比值与空白组比较有显著性差异(p<0.01),24h时ros比值为2.56,细胞损伤明显且稳定,故本实验诱导模型选择浓度为300μmol/lh2o2溶液诱导huvec细胞24h。
表3h2o2诱导细胞不同时间后对细胞半数抑制浓度的影响
表4不同浓度h2o2处理细胞不同时间对细胞内ros水平的影响
注:与空白组(0μmol/l)比较,*代表有显著性差异(p<0.05),**代表有极显著性差异(p<0.01)。
实施例1~3对h2o2诱导huvec细胞氧化损伤的影响:取对数生长期细胞传代后随机分组进行试验。包括空白对照组,h2o2诱导模型组,咖啡酸对照组,花翠素-3-o-葡萄糖苷组(10μmol/l、50μmol/l、100μmol/l),实施例1组(10μmol/l、50μmol/l、100μmol/l),实施例2组(10μmol/l、50μmol/l、100μmol/l),实施例3组(10μmol/l、50μmol/l、100μmol/l)。按照上述方法分别测定细胞增殖率及细胞内ros水平。结果如表5和表6所示,h2o2诱导模型组的细胞增殖率为空白对照组的0.52倍,具有极显著性差异(p<0.01),表明建模成功,同时细胞内ros释放量显著增加(p<0.01)。不同浓度的咖啡酸对照组与h2o2诱导模型组相比,细胞增殖率无显著性差异(p>0.05),说明浓度为100μmol/l的咖啡酸对氧化损伤细胞处理24h时无保护作用;不同浓度的花翠素-3-o-葡萄糖苷组处理后,细胞增殖率显著增加,浓度在50μmol/l与100μmol/l时,与h2o2诱导模型组相比有显著性差异(p<0.05)和极显著性差异(p<0.01),细胞内ros释放量显著降低(p<0.01),说明花翠素-3-o-葡萄糖苷组对h2o2诱导huvec细胞氧化损伤具有一定的保护作用,且呈浓度依赖性。不同浓度的实施例组处理后,细胞增殖率显著增加,浓度在50μmol/l与100μmol/l时,与h2o2诱导模型组相比均出现极显著性差异(p<0.01),同时细胞内ros释放量显著降低(p<0.01),与花翠素-3-o-葡萄糖苷组相比,细胞增殖率均出现极显著性差异(p<0.01),ros水平无统计学差异(p>0.05)。说明实施例制得的花翠素-3-o-(6"-o-咖啡酰基)-葡萄糖苷,不仅对h2o2诱导huvec细胞氧化损伤具有一定的保护作用,同时其抗氧化活性比花翠素-3-o-葡萄糖苷强,且呈浓度依赖性。
表5不同浓度的实施例的衍生物对huvec细胞增殖率的影响
注:与空白组比较,*代表有显著性差异(p<0.05),**代表有极显著性差异(p<0.01);与模型组比较,δ代表有显著性差异(p<0.05),δδ代表有极显著性差异(p<0.01);与未稳定化花青素组比较,▲代表有显著性差异(p<0.05),▲▲代表有极显著性差异(p<0.01)。
表6不同浓度的实施例的衍生物对huvec细胞内ros水平的影响
注:与空白组比较,*代表有显著性差异(p<0.05),**代表有极显著性差异(p<0.01);与模型组比较,δ代表有显著性差异(p<0.05),δδ代表有极显著性差异(p<0.01);与未稳定化花青素组比较,▲代表有显著性差异(p<0.05),▲▲代表有极显著性差异(p<0.01)。
1.4对实施例1~3制备的衍生物进行抗肿瘤活性试验。
试验采用细胞试验,细胞培养液和缓冲液的配制:溶液1:完全dmem培养液-10,移取fbs50ml、100×neaa溶液5ml(10mmol/l)、100×双抗5ml(10,000u/ml青霉素和10,000g/ml链霉素),加不完全dmem培养液至500ml;ph7.35。溶液2:完全rpmi培养液-10,移取fbs100ml、100×neaa溶液5ml(10mmol/l)、100×双抗5ml(10,000u/ml青霉素和10,000g/ml链霉素),加不完全rpmi培养液至500ml;ph7.35。肿瘤细胞的培养:hepg-2肝癌细胞采用溶液1培养,hl-60急性早幼粒白血病细胞采用溶液2培养。从液氮中分别取出上述两种细胞,分别以对应溶液复苏,每天换液,待细胞汇合率约80%时传代,传代比例为1:3。待细胞稳定后继续培养1~2代。
实施例1~3抗肿瘤活性的考察:
取传代培养3~4代后细胞进行抗肿瘤活性的筛选,待细胞生长至80%左右,用0.25%胰蛋白酶-edta将细胞从培养瓶瓶壁上消化下来,离心后加入对应溶液将细胞稀释到2×105cells/ml,充分吹打均匀。
将细胞悬液接种于96孔板中,每孔100μl,置于co2培养箱中37℃培养24h。迅速吸出培养液,加入含有不同浓度药物的培养基100μl,再次置于co2培养箱作用48h。其中药物浓度分别为100μm、50μm、25μm、12.5μm、6.25μm、3.125μm。48h后取出96孔板,每孔加入cck-8溶液10μl,轻微振摇使混合均匀,置于co2培养箱中37℃培养1~4h,结果稳定后进行od值的测定,波长为450nm。每个96孔板设6个空白孔作为对照,每个化合物每种肿瘤细胞平行进行三次实验,所有结果进行统计分析。结果如表7和表8所示,实施例1~3制得的花翠素-3-o-(6"-o-咖啡酰基)-葡萄糖苷抑制hepg-2细胞生长的ic50浓度分别为63.25μm/l,58.21μm/l,60.38μm/l,抑制hl-60细胞生长的ic50浓度分别为49.32μm/l,48.25μm/l,47.67μm/l,具有较好的抗肿瘤活性。
表7不同实施例的衍生物抗hepg-2活性结果
表8不同实施例的衍生物抗hl-60活性结果
对所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
- 还没有人留言评论。精彩留言会获得点赞!