新型EED-EZH2相互作用小分子抑制剂的确定和评价的制作方法
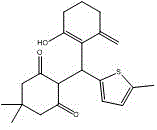
本发明属于医药领域,涉及图1所示的化合物35作为eed-ezh2相互作用的小分子抑制剂的用途及其在制备预防和/或治疗恶性淋巴瘤的药物中的用途。
背景技术:
组蛋白的甲基化是介导多种基本细胞过程的重要表观遗传修饰之一。多梳抑制复合物2(polycombrepressivecomplex2,即prc2)具有组蛋白甲基转移酶活性,可催化组蛋白h3第27位赖氨酸的三甲基化,在胚胎发育、干细胞可塑性和细胞分化等多种生物学进程中发挥重要的调节作用。prc2由e2h2、eed和suz12三个关键亚基构成,e2h2是催化亚基,与eed和suz12形成复合物后发挥作用,eed或suz12的缺失都会使prc2失活。
大量研究表明,prc2核心组分的活性异常与多种疾病密切相关,尤其是恶性肿瘤的侵袭、发生和发展。在多种人类癌症中,如乳腺癌、前列腺癌、肝癌、大肠癌、神经胶质瘤、淋巴瘤,ezh2、eed和suz12表达水平显著升高,而ezh2的下调可以显著抑制这些肿瘤细胞的增殖。因此,prc2是一种非常有前景的抗肿瘤靶点。
靶向ezh2或eed-ezh2蛋白-蛋白相互作用(ppi)是目前研发prc2抑制剂的两种策略。在已报道的ezh2抑制剂中,gsk126、epz6438和cpi-1205已进入临床试验。然而,一方面,由于ezh2抑制剂直接作用于其天然底物s-腺苷甲硫氨酸(sam)的结合位点,而人体内超过50种甲基转移酶中都有类似的sam结合口袋,因而,需要花费大量的实验资源去检测化合物对其他同源蛋白的选择性;另一方面,ezh2需要与eed和suz12形成复合物后才能发挥催化活性,因而,通过抑制ezh2与其辅助蛋白(如eed)的相互作用也可以抑制ezh2的催化活性,同时,由于ezh2蛋白-辅助蛋白相互作用位点的特异性远高于其与sam结合位点的特异性,故靶向蛋白-蛋白相互作用的位点抑制剂可以解决该化合物的选择性难题。2013年,哈佛医学院的woojinkim等人根据ezh2与eed的复合物晶体结构,设计合成了一种ezh2环肽类化合物(stapledpeptide),选择性抑制ezh2与eed的结合,促进prc2多个组分的降解,进而抑制其甲基转移酶活性。相比ezh2抑制剂,该环肽类化合物不仅对依赖ezh2催化活性的肿瘤细胞有杀伤作用,更重要的是,对一些虽然依赖ezh2但是不依赖其甲基转移酶活性的肿瘤细胞(如基底样恶性乳腺癌、雄激素抵抗性恶性前列腺癌等)具有明显的增殖抑制作用,因而,极大地扩大了prc2复合物抑制剂的应用范围。然而,由于多肽类化合物通常存在细胞渗透能力低、代谢稳定性差和制备昂贵等缺点,其在临床上的使用受到了严重的制约。与此相反,小分子化合物具有通透性好、代谢稳定性高和制备难度低等特点,有效弥补了多肽类化合物的不足。因此,特异性靶向eed-ezh2相互作用的高活性、高成药性小分子抑制剂具有更广阔的临床应用前景,可以为多种依赖于ezh2的恶性肿瘤的治疗提供有力的理论支撑。
eed与ezh2的相互作用界面(顶部和底部)均可以作为ppi抑制剂的靶点。诺华制药公司的研究员已经发现了多个靶向eed顶部的ppi抑制剂并确定了共晶复合物结构,而目前,靶向eed底部的ppi抑制剂却很少。
综上所示,本领域迫切需要开发靶向eed底部的ppi小分子抑制剂。
技术实现要素:
本发明的发明人为解决上述难题,通过虚拟筛选结合生物学评价,发现了一种靶向eed底部的新型eed-ezh2相互作用小分子抑制剂,并测定了其对弥漫性大b细胞淋巴瘤细胞系toledo细胞的抗增殖活性。由此本发明的一个目的在于提供一种新型eed-ezh2相互作用小分子抑制剂——化合物35,如图1所示。
本发明的另一个目的在于提供图1所示的化合物35在制备预防和/或治疗恶性淋巴瘤的药物中的用途。
优选地,所述恶性淋巴瘤为弥漫性大b细胞淋巴瘤相关的疾病。
图1所示的化合物35作为eed-ezh2相互作用的抑制剂和prc2的抑制剂,能够有效地抑制eed-ezh2相互作用,从而抑制prc2的催化活性,进而有效地预防和治疗与弥漫性大b细胞淋巴瘤相关的恶性淋巴瘤疾病。
附图说明
图1新型eed-ezh2相互作用小分子抑制剂——化合物35。
图2虚拟筛选流程图。
图3化合物35的预测结合模式。
图4maccs指纹图谱相似度。
图5化合物35的化学结构和抑制曲线。
图6化合物35的抗细胞增殖活性。
具体实施方式
在本发明中,采用荧光偏振技术对候选化合物进行高通量筛选,该荧光偏振方法已被广泛地用于蛋白质-蛋白质或核酸-蛋白质之间相互作用的小分子抑制剂的高通量筛选工作,其方法是在缓冲液中加入eed蛋白、fitc-ezh2多肽示踪底物和待检测化合物,室温孵育数小时后,用酶标仪检测其荧光偏振值,结果表明,化合物35可以与ezh2多肽竞争结合eed蛋白;采用分子对接技术探讨了化合物35与eed的结合方式;进行体外细胞活性分析,测定化合物35对弥漫性大b细胞淋巴瘤细胞系的toledo细胞的抗增殖活性,结果表明,化合物35对toledo细胞表现出剂量依赖性抗增殖活性。
下面结合具体实施例对本发明作进一步阐述,这些实施例并非限制性的,而仅作为示例说明本发明。
实验例1:对specs数据库进行基于分子对接的虚拟筛选,选出候选化合物,采用分子对接技术预测苗头化合物与eed的结合模式。
在之前的研究中,我们通过虚拟筛选发现了第一个靶向eed底部的有效eed-ezh2相互作用抑制剂(阿司咪唑ic50=93.80μm),为了获得更多靶向eed底部的ppi抑制剂,对包含211838个化合物的specs数据库进行基于分子对接的虚拟筛选。
分子对接使用软件包maestro7.5.中的glide5.5程序。使用蛋白制备向导工作流程中的默认设置最小化蛋白质坐标,以分解结合自由能确定的关键残基(r46残基)为中心,构建一个长度为15å的立方体盒子作为结合位点,同时在该盒子范围内生成格点;将specs数据库准备好的化合物以标准精度(sp)模式导入预先设定好的结合位点中,对排名前500的化合物进行进一步的对接验证和额外精度(xp)分析;随后,采用pipelinepilot7.5对排名前200的化合物进行聚类分析,根据聚类分析结果,选择50种候选化合物并从specs数据库供应商购买;之后采用荧光偏振实验检测它们的eed-ehz2相互作用抑制活性,选出苗头化合物,虚拟筛选流程图如图2所示。
预测的化合物35的结合模式如图3所示,从图中可知,化合物35能很好地占据r46的结合位点,与残基l315、g316、d317、l318、w373、q374、l391、v393和p396形成疏水作用。
实施例2:用rdkit计算maccs指纹图谱相似度。
采用molfromsmiles将化合物35和参考化合物(阿司咪唑和盐酸阿扑吗啡)的smiles格式转换成二维结构,计算maccs关键指纹图谱相似度并做比较。实验结果如图4所示,从结果可以看出,化合物35与阿司咪唑和盐酸阿扑吗啡的结构相似度很小。
实施例3:蛋白质的表达和纯化。
将eed基因(编码氨基酸序列为81~441的eed蛋白)亚克隆到一个修饰过的pet28a载体(n-末端有6×hissumo标记)中,用大肠杆菌菌株bl21(de3)在16℃下过度表达重组蛋白,收集菌株,超声,离心(18000rpm,30min,4℃),取上清液;用镍亲和层析色谱(gehealthcare)和尺寸排阻色谱(superdex75;gehealthcare)纯化蛋白质。蛋白质储存在含有25mmhepes(ph8.0),150mmnacland1mmdtt的缓冲液中。
实施例4:荧光偏振方法(fluorescencepolarization,fp)检测候选化合物对eed-ezh2相互作用的影响。
荧光偏振实验,采用异硫氰酸荧光素(fluoresceinisothiocyanate,fitc)标记的ehz2多肽作为示踪底物,从100mmdmso储存液中制备一系列稀释浓度的待测化合物35,在40μlfp缓冲液(25mmhepes,ph8.0,150mmnacl,0.1mg/mlbsa和0.01%np40)中加入625nmeed蛋白、20nmfitc-ezh2多肽示踪底物(残基为39-63)和不同浓度的待测化合物35,室温孵育2小时,用全自动多功能酶标仪(polarstaromegamicroplatereader,envision,perkinelmer)检测荧光偏振值,所用激发光和发射光波长分别为485nm和520nm;每组均设三组重复实验,实验数据采用graphpadprism6.0软件分析并制图。结果如图5所示,化合物35(ic50=63.88±6.48μm)表现出比阿司咪唑(ic50=93.80μm)更有效的抑制活性,表明化合物35可以与ezh2多肽竞争结合eed蛋白,表现出有效的抑制活性。
实施例5:体外细胞活性分析实验检验化合物35对prc2活性失调引起的dlbcls癌细胞增殖的影响。
在含有10%胎牛血清(fbs)和1%青霉素/链霉素的rpmi1640培养基上,以每孔2x104个细胞的密度培养指数生长的细胞,然后用100μm和50μm的化合物35进行孵育,孵育三天后,用celltiter-gloluminescentcellviabilitykit(promega)检测细胞活性,实验结果如图6所示,化合物35对toledo细胞表现出剂量依赖性抗增殖活性。
综合上述具体实施例测定结果,化合物35能很好地占据r49的结合位点,与两个已知活性化合物(阿司咪唑和盐酸阿扑吗啡)的指纹图谱相似性很小,表现出比阿司咪唑(ic50=93.80μm)更有效的抑制活性,对toledo细胞表现出剂量依赖性抗增殖活性。我们的工作为发现更多prc2抑制剂提供了新的骨架,为设计新型eed-ezh2相互作用抑制剂提供了结构上的启示。
- 还没有人留言评论。精彩留言会获得点赞!