一种制备香草乙酮的方法及其应用与流程
本发明涉及一种制备香草乙酮的方法及其应用。
背景技术:
香草乙酮又称香荚兰乙酮,乙酰香草酮,具有弱香草嗅味,不仅用于香料工业也是重要的有机合成原料,可用于合成精神类药物伊潘立酮和多种新型抗疟药。香草乙酮同时也是一种常用的抗氧化剂,是目前公认的特异性烟酰胺腺嘌呤二核苷酸氧化酶抑制剂,在医学上还常用作强心剂和利尿药。
鉴于香草乙酮在化工和医药行业的重要作用,近年来有许多制备香草乙酮的研究报道。譬如,邓日玲等人报道的利用石化产品愈创木酚钠(邻甲氧基苯酚钠)为原料制备香草乙酮,其工艺步骤繁琐,条件较为苛刻而且产率也相对较低(《纤维素科学与技术》2009,17(2):27-33)。周永红等人报道利用硝基苯化合物作为氧化剂氧化木质素类化合物制备香草乙酮的方法。该方法需要使用对硝基苯甲酸或3-硝基苯甲酸等作氧化剂,反应温度较高(160℃~200℃),后处理繁琐,收率低,成本高(cn102295547)。黄伟彬等人报道了愈创木酚与溴代正丙烷经丙基化反应得1-正丙氧基-2-甲氧基苯,1-正丙氧基-2-甲氧基苯与醋酐经傅-克乙酰基化反应得3-甲氧基-4-正丙氧基苯乙酮和3-丙氧基-4-甲氧基苯乙酮的混合物,随后以无水三氯化铝对3-甲氧基-4-正丙氧基苯乙酮和3-丙氧基-4-甲氧基苯乙酮混合物进行选择性脱丙基反应制备合成香草乙酮和异香草乙酮(《合成化学》2013,21(4):472-475)。该方法步骤复杂,成本高且用到易制毒的管制原料醋酐,不利于大规模生产,其最大的缺点在于选择性差,在合成香草乙酮的同时并发生成异香草乙酮,原子经济性差。
上述报道工艺都或多或少存在技术上缺陷,如反应条件较为苛刻,需要较高反应温度,对工艺设备要求较高同时会造成环境污染,反应选择性差,在生产目标产物同时有别的副产物并发产生等。
另一方面,香草乙酮作为非典型精神病药物伊潘立酮关键中间体,如us4366162中以香草乙酮为起始物料,与1-溴-3氯丙烷反应的中间体后,再与化合物e反应形成伊潘立酮(式i),
鉴于此,药化研究人员急需开发一种新的、良好环境友好性且工艺可操作性强、收率高的香草乙酮的制备方法。
技术实现要素:
本发明提供一种新的制备香草乙酮的方法,包括将乙酸2-甲氧基苯酚酯加入路易斯酸和硝基苯中反应的步骤,进一步还包括反应完毕后冷却,加入盐酸溶液,搅拌溶清的步骤。该工艺方法良好环境友好性且工艺可操作性强、收率高的,便于工业化生产。
在一些实施方案中,反应温度选自约50~80℃,包括但不限于50℃、55℃、60℃、65℃、70℃、75℃、80℃或任意两数值之间任意值。在具体方案中,反应温度直接关系到反应速率,进而影响香草乙酮生成量,以及相应副产物的生成量,如异香草乙酮和/或乙酸2-甲氧基苯酚酯水解产物。另一方面,反应温度过高,反应体系中会出现黑色絮状物,推测可能源于具有酚羟基结构化合物的氧化,
在一些实施方案中,所述反应时间选自约7~10小时,包括但不限于7h、8h、9h、10h或任意两数值之间任意值。在具体方案中,反应时间影响反应的收率,反应时间过短,反应过程2-甲氧基苯酚酯水解产物会大量出现,且原料转化不完全。
另一方面,一些实施方案提供的方法中所述路易斯酸选自氯化锌、氯化铝、三氯化铁、三溴化铁、四氯化锡、四氯化钛和多聚磷酸的中至少一种,优选氯化铝。为了确保反应顺利进行,一些实施方案提供的方法中乙酸2-甲氧基苯酚酯与路易斯酸的投料摩尔比约为1:1.6~1:2.5,包括但不限于1:1.6、1:1.7、1:1.8、1:1.9、1:2.0、1:2.1、1:2.2或任意两数值之间任意值,优选约1:2.0~1:2.5。
进一步地,本发明中所述方法中乙酸2-甲氧基苯酚酯由愈创木酚和乙酰氯反应形成,其中愈创木酚与乙酰氯投料摩尔比为约1:1.1~1:3,包括但不限于1:1.1、1:1.2、1:1.3、1:1.4、1:1.5、1:1.6、1:1.7、1:1.8、1:1.9、1:2.0、1:2.2、1:2.4、1:2.6、1:2.8、1:3.0或任意两数值之间任意值,优选约1:2~1:3。
进一步地,在一些实施方案中,所述愈创木酚和乙酰氯反应温度选自约-5~25℃,包括但不限于-5℃、0℃、5℃、10℃、15℃、20℃、25℃或任意两数值之间任意值,优选约15~25℃,更优选约25℃。另一方面,所述愈创木酚和乙酰氯反应时间选自约1~6h,包括但不限于1、2、3、4、5、6h或任意两数值之间任意值,优选约4~6h,更优选6h。
为了确保酰胺化反应的进行,愈创木酚和乙酰氯在缚酸剂条件下反应,所述缚酸剂选自三乙胺、吡啶、4-二甲氨基吡啶、n,n-二甲基甲酰胺、n,n-二异丙基乙胺或四甲基乙二胺的中至少一种,优选三乙胺。
进一步地,本发明所述制备香草乙酮的方法中还包括有机溶剂萃取、干燥、浓缩或重结晶步骤中的至少一种。
在一些实施方案中,用于萃取反应的有机溶剂选自二氯甲烷、1,2-二氯乙烷、氯仿。
在一些实施方案中,用于重结晶的溶剂选自丙酮、乙醇、异丙醇、乙酸乙酯、石油醚或甲苯中的任一种或多种任意比的混合溶剂。
在一些实施方案中,本发明制备香草乙酮的方法包括:
1)愈创木酚和乙酰氯反应形成乙酸2-甲氧基苯酚酯的步骤,其中愈创木酚与乙酰氯投料摩尔比为1:1.1~1:3,优选1:2~1:3;所述反应温度优选-5~25℃,更优选25℃;所述反应时间优选1~6h,更优选6h;
2)将乙酸2-甲氧基苯酚酯加入路易斯酸和硝基苯中反应的步骤,进一步还包括反应完毕后冷却,加入盐酸溶液,搅拌溶清的步骤,所述反应温度优选50~80℃;所述反应时间优选7~10小时。
3)有机溶剂萃取、干燥、浓缩或/和重结晶步骤,其中用于萃取的有机溶剂选自二氯甲烷、1,2-二氯乙烷、氯仿;用于重结晶的溶剂选自丙酮、乙醇、异丙醇、乙酸乙酯、石油醚或甲苯中的任一种或多种任意比的混合溶剂。
在另一些实施方案中,本发明制备香草乙酮的方法包括:
1)将愈创木酚(1当量),三乙胺(2当量)和乙酰氯(1.5当量)溶解于1,2-二氯乙烷中,在15-25℃下反应,用0.5mol/l盐酸调节体系至酸性,再用1,2-二氯乙烷萃取,浓缩后再精馏得到2-甲氧基乙酸苯酯。
2)把氯化铝加入硝基苯中搅拌均匀,滴加2-甲氧基乙酸苯酯,在80℃下反应,体系冷却至室温,加浓盐酸酸化;
3)萃取,萃取液减压浓缩,重结晶得香草乙酮。
另一方面,本发明还提供一种制备式i化合物方法,包括前述的方法制备香草乙酮的步骤,以及香草乙酮转化为i化合物的步骤,
在一些实施方案中,制备式i化合物方法还包括在碱性条件下,香草乙酮与化合物b反应形成式i化合物的步骤,
其中,x选自离去基团,优选卤素(包括氟、氯、溴或碘)、ots、oms,更优选氯或ots;所述碱选自三乙铵或碳酸钾。
在一些实施方案中,制备式i化合物方法还包括在碱性条件下,其还包括在碱性条件下,香草乙酮与化合物c反应形成化合物d的步骤,以及化合物d与化合物e反应形成式i化合物的步骤,
其中,x选自离去基团,优选卤素(包括氟、氯、溴或碘)、ots、oms,更优选氯或ots;所述碱选自三乙铵或碳酸钾。
进一步地,制备式i化合物方法具体反应参照于cn102212063a,并将相关内容引入本发明中以说明。
本发明所用术语“约”是指数值在由本领域一般技术人员所测定的具体值的可接受误差范围内,所述数值部分取决于怎样测量或测定(即测量体系的限度)。例如,在本领域每一次实行中“约”可意味着在1内或超过1的标准差。或者,“约”或“基本上包含”可意味着至多20%的范围。此外,特别对于生物学系统或过程而言,该术语可意味着至多一个数量级或数值的至多5倍。除非另外说明,否则当具体值在本发明和权利要求中出现时,“约”或“基本上包含”的含义应该假定为在该具体值的可接受误差范围内。
化合物的结构可通过核磁共振(nmr)或/和质谱(ms)来确定的。nmr位移(δ)以10-6(ppm)的单位给出。nmr的测定是用brukeravance-400核磁仪,测定溶剂可为氘代二甲基亚砜(dmso-d6)、氘代氯仿(cdcl3)、氘代甲醇(cd3od),内标为四甲基硅烷(tms);ms的测定用finniganlcqad(esi)质谱仪(生产商:thermo,型号:finniganlcqadvantagemax)。
hplc的测定可使用安捷伦1200dad高压液相色谱仪或waters2695-2996高压液相色谱仪。
附图说明
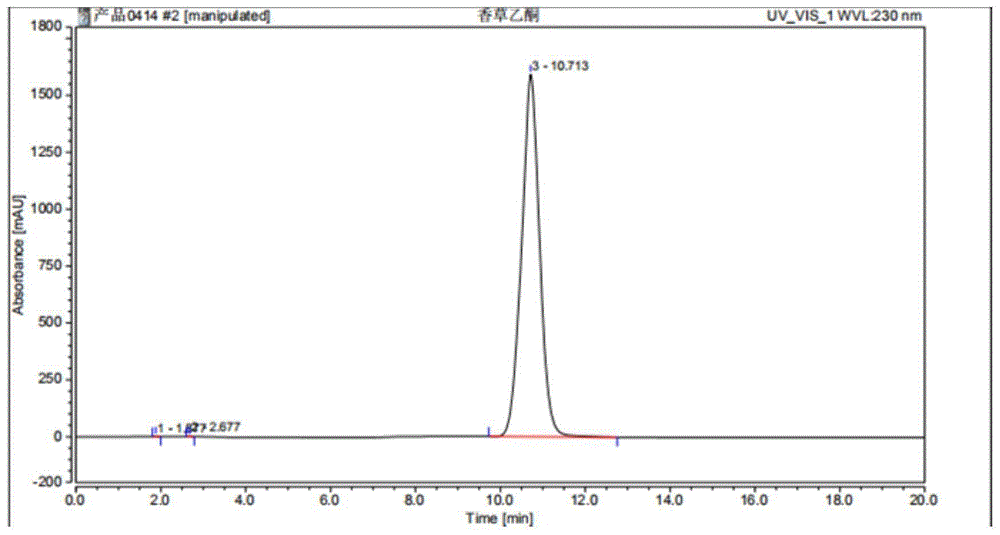
图1:香草乙酮高效液相谱图,其高效液相谱图显示保留时间为10.713,纯度为99.63%。
具体实施方式
实施例1:
将愈创木酚(200.00g,1.61mol),吡啶(254.70g,3.22mol)和乙酰氯(189.58g,2.42mol)溶解于500ml的1,2-二氯乙烷中,在-5℃下反应6h。反应结束后,用0.5mol/l盐酸调节体系至ph=3,再用250ml1,2-二氯乙烷萃取,浓缩后再精馏得到2-甲氧基乙酸苯酯。经液相色谱检测纯度为95%,收率92%。
实施例2:
参照实施例1中的方法制备2-甲氧基乙酸苯酯,筛选不同的反应体系以便获得最优的反应条件,具体数据如下:
注:a表示缚酸剂为三乙胺,b表示缚酸剂为吡啶,c表示缚酸剂为n,n-二甲基甲酰胺,d表示缚酸剂为4-二甲氨基吡啶。
结论:当愈创木酚与缚酸剂三乙胺摩尔比为1:3,25℃反应6h时,所得2-甲氧基乙酸苯酯的收率最优,纯度更好。当然其他收率为90%以上的也是可以接受的。
实施例3:
将愈创木酚(200.00g,1.61mol),三乙胺(325.83g,3.22mol)和乙酰氯(189.58g,2.42mol)溶解于500ml的1,2-二氯乙烷中,在25℃下反应6h。反应结束后,用0.5mol/l盐酸调节体系至ph=3,再用250ml1,2-二氯乙烷萃取,浓缩后再精馏得到2-甲氧基乙酸苯酯。经液相色谱检测纯度为98%,收率96%。
实施例4:
把氯化铁(9.73g,0.06mol)加入25ml硝基苯中搅拌均匀,滴加2-甲氧基乙酸苯酯(5.0g,0.03mol),在60℃下反应8h,体系冷却至室温,加入浓盐酸至体系澄清,用25ml1,2-二氯乙烷萃取,合并有机相。有机相用25ml2mol/l氢氧化钠洗涤,收集水相。水相用浓盐酸酸化至ph=2~3,然后再用1,2-二氯乙烷20ml萃取,合并有机相,减压浓缩,粗品使用10ml乙醇重结晶。经液相色谱检测纯度为99%,收率55%。
熔点为112~114℃。
1hnmrδ:7.51~7.54(m,2h,arh),δ:6.92~6.95(d,j=12hz,1h,arh),δ:6.12(s,1h,oh),δ:3.94(s,3h,ch3),δ:2.55(s,3h,ch3)。
13cnmrδ:196.85,150.42,146.63,130.23,124.03,113.78,109.72,56.08,26.11。
hr-esi-msm/z:[m+h]+=167.0536。
实施例5
参照实施例4中的方法制备香草乙酮,筛选不同的反应体系以便获得最优的反应条件,具体数据如下:
注:图表中a表示为2-甲氧基乙酸苯酯;na表示为未反应。
结论:同等反应条件下,筛选不同的路易斯酸,当选用氯化铝时,目标产物转化率最高,达到67.08%。
实施例6
把氯化铝(10.00g,0.075mol)加入25ml硝基苯中搅拌均匀,滴加2-甲氧基乙酸苯酯(5.0g,0.03mol),在80℃下反应8h,体系冷却至室温,加入浓盐酸至体系澄清,用25ml1,2-二氯乙烷萃取,合并有机相。有机相用25ml2mol/l氢氧化钠洗涤,收集水相。水相用浓盐酸酸化至ph=2~3,然后再用1,2-二氯乙烷20ml萃取,合并有机相,萃取液减压浓缩,粗品使用10ml乙醇重结晶。经液相色谱检测分离纯度为99%,收率63%。
实施例7
参照实施例6中的方法制备香草乙酮,筛选不同的反应体系以便获得最优的反应条件,具体数据如下:
结论:异构体随温度升高在逐渐减少,但是温度越高,体系中黑色絮状物越多,后处理分层越困难。
实施例8
参照实施例6中的方法制备香草乙酮,筛选不同的反应体系以便获得最优的反应条件,具体数据如下:
结论:其他反应条件不变,2-甲氧基苯酚酯与氯化铝的投料摩尔比为1:2.5时,目标产物转化率最高(84.15%),继续增加氯化铝用量,目标物转化率又下降同时异构体增加。
实施例9
参照实施例6中的方法制备香草乙酮,筛选不同的反应体系以便获得最优的反应条件,具体数据如下:
结论:其他反应条件不变,反应时间为8h时,目标产物转化率最高(80.92%),但随反应时间延长,目标物转化率又下降。
实施例10
把氯化铝(10.00g,0.075mol)加入25ml硝基苯中搅拌均匀,滴加2-甲氧基乙酸苯酯(5.0g,0.03mol),在80℃下反应8h,体系冷却至室温,加入浓盐酸至体系澄清,用25ml1,2-二氯乙烷萃取,合并有机相。有机相用25ml2mol/l氢氧化钠洗涤,收集水相。水相用浓盐酸酸化至ph=2~3,然后再用1,2-二氯乙烷20ml萃取,合并有机相。萃取液减压浓缩,粗品使用10ml乙酸乙酯重结晶。经液相色谱检测纯度为99%,收率78%。
实施例11
把氯化铝(20.06kg,150.43mol)加入40l硝基苯中搅拌均匀,滴加2-甲氧基乙酸苯酯(10.00kg,60.18mol),在80℃下反应8h,体系冷却至室温,加入浓盐酸至体系澄清,用35l1,2-二氯乙烷萃取,合并有机相。有机相用43l2mol/l氢氧化钠洗涤,收集水相。水相用浓盐酸酸化至ph=2~3,然后再用1,2-二氯乙烷40l萃取。萃取液减压浓缩,粗品使用20l乙酸乙酯重结晶。经液相色谱检测纯度为99%,收率82%。
- 还没有人留言评论。精彩留言会获得点赞!