一种烯烃硫醚类订书肽及其制备方法与应用与流程
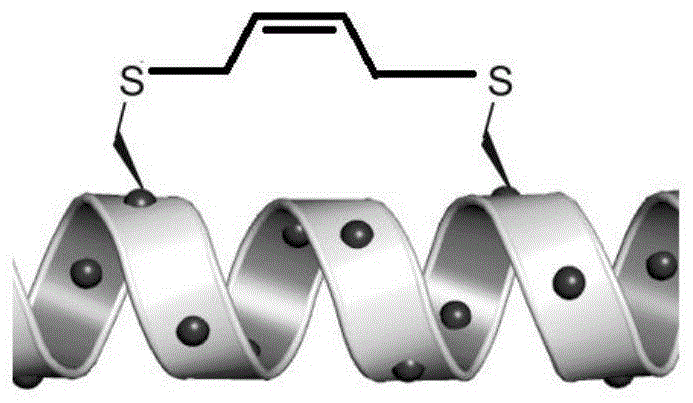
本发明属于医药领域,具体涉及一种烯烃硫醚类订书肽及其制备方法与应用。技术背景肽类分子因具有活性高、毒性低、用药剂量低等优点,在药物研发中受到广泛的关注。但是肽类分子能够作为多肽类药物实现上市销售的很少,主要原因是多肽类药物多为线性多肽,因空间构象不固定具有透细胞膜性能差和体内易分解的缺点。多肽类药物空间构象不固定的原因在于:α-螺旋是蛋白质中最为普遍的二级结构单位,且在蛋白与蛋白、蛋白与dna的相互作用中发挥极其重要的作用。如果从蛋白质中截取该α-螺旋结构合成得到短肽片段,该短肽片段在水溶液中不能保持稳定的α-螺旋二级结构,从而具有空间构象不固定的缺点。因此人们不断尝试能够保持多肽α-螺旋结构稳定的方法,2000年verdine等人用碳碳键作为连接桥(订书桥)来稳定多肽中的α-螺旋结构,得到的多肽称为订书肽(stapledpeptides),即:在固相合成多肽链的过程中引入两个α-甲基-α-烯基非天然氨基酸,然后通过烯烃复分解反应实现环合,使得线性多肽的构象得以固定,得到订书肽。与线性肽相比,订书肽因构象固定,与靶标通常具有更好的亲和力,也具有良好的透膜性能、体内稳定性和较长的体内半衰期。然而,α-甲基-α-烯基氨基酸(如(s)-2-氨基-2-甲基-4-戊烯酸)是非天然氨基酸,是目前制备订书肽的重要中间体。由于其具有手性中心,需要手性合成或通过拆分制备,且在合成订书肽时需要在氨基酸的α位引入侧链烯烃以形成α位全取代的结构,这就要求该α-甲基-α-烯基氨基酸本身必须具有高的光学纯度,否则合成出来的订书肽会出现非对映异构体的杂质,造成后期分离纯化的困难。目前通过手性合成或拆分的办法制备光学纯度高的α-甲基-α-烯基非天然氨基酸成本很高,也就限制了此类订书肽的推广和应用。技术实现要素:本发明的目的是针对现有技术中存在的技术缺陷,第一方面,提供一种烯烃硫醚类订书肽,其结构式见式(ⅰ),其中,x为n个天然氨基酸组成的多肽,n为2-4的整数,m1和m2分别为1-3数值范围内的整数,c为半胱氨酸中的α-碳原子,y1为3-8个天然氨基酸组成的多肽;优选的,y2为2-5个天然氨基酸组成的多肽或为天然氨基酸中的任意一种。所述天然氨基酸为甘氨酸(g)、丙氨酸(a)、缬氨酸(v)、亮氨酸(l)、异亮氨酸(i)、苯丙氨酸(f)、脯氨酸(p)、色氨酸(w)、丝氨酸(s)、酪氨酸(y)、半胱氨酸(c)、蛋氨酸(m)、天冬酰胺(n)、谷氨酰胺(q)、苏氨酸(t)、天冬氨酸(d)、谷氨酸(e)、赖氨酸(k)、精氨酸(r)、组氨酸(h)。y1选自亮氨酸(l)、赖氨酸(k)、谷氨酰胺(q)、脯氨酸(p)、苏氨酸(t)、苯丙氨酸(f)、丝氨酸(s)、天冬氨酸(d)中的至少三种;y2选自亮氨酸(l)、赖氨酸(k)、谷氨酰胺(q)、脯氨酸(p)中的至少两种;x为赖氨酸(k)、色氨酸(w)、亮氨酸(l)、苯丙氨酸(f)、天冬氨酸(d)、丝氨酸(s)中的一种或多种组成的含有n个氨基酸的多肽。当y1为6-8个天然氨基酸组成的多肽时,y2为2-4个天然氨基酸组成的多肽或为所述天然氨基酸中的任意一种;优选的,y1的序列结构由ho-向nh2-顺序为q-p-t-f-s-d时,y2的序列结构由ho-向nh2-顺序为l-p或为所述天然氨基酸中的任意一种。当y1为3-5个天然氨基酸组成的多肽时,y2为2-5个天然氨基酸组成的多肽;优选的,y1的序列结构由ho-向nh2-顺序为l-k-l时,y2的序列结构由ho-向nh2-顺序为l-q。m1=1或2,m2=1或2。其结构式为式(1)-式(12)中的一种:第二方面,本发明提供一种药物组合物,其活性成分为上述烯烃硫醚类订书肽。第三方面,本发明提供一种制备上述烯烃硫醚类订书肽的方法,,在半胱氨酸的巯基上引入烯烃链得到非天然氨基酸fmoc-cys(c3-c5-ene)-oh作为订书肽环合关键砌块,通过烯烃复分解反应进行环合。包括以下步骤:(1)将半胱氨酸在碱的作用下与卤代烯烃(优选c3-c5的卤代烯烃)反应,再经fmoc保护制得烯烃醚化的非天然氨基酸fmoc-cys(c3-c5-ene)-oh;(2)使用wang树脂通过标准固相多肽合成方法组装线性多肽,且在多肽序列组装过程中在x多肽位置的之前和之后分别加入所述非天然氨基酸fmoc-cys(c3-c5-ene)-oh;(3)步骤(2)得到的线性多肽组装完毕后,加入grubbs催化剂,通过烯烃复分解反应进行环合。具体的,还可包括常规步骤:反应结束后使用三氟乙酸裂解液切割,得到订书肽粗品;高压制备液相色谱分离纯化,冷冻干燥得到所述烯烃硫醚类订书肽。第三方面,本发明提供上述烯烃硫醚类订书肽在制备抗肿瘤药物中的应用,所述肿瘤包括但不限于乳腺癌和结肠癌。本发明提出的烯烃硫醚类订书肽是一种新的订书肽桥连结构,制备时在手性的半胱氨酸的α-碳原子(氨基酸中与羧基相连的c为α-碳原子)的巯基上通过硫醚化,引入烯烃链,再将两个烯烃硫醚化的半胱氨酸插入肽链特定位置中,通过烯烃复分解反应环合得到含有烯烃硫醚链的订书肽。这种订书肽合成方便,且用光学纯度高的烯烃硫醚化半胱氨酸作原料即可;另外由于烯烃连接在半胱氨酸远端的巯基上,对肽链骨架构象的影响较小,不影响肽链本身的功效。此外,本发明的烯烃硫醚类订书肽因构象能够保持固定,在保留线性肽与靶标具有更好的亲和力、良好的透膜性能、体内稳定性和较长的体内半衰期等优点的前提下,还具有明显的抗肿瘤活性,尤其抗乳腺癌(如人乳腺癌细胞mcf-7)、抗结肠癌(人结肠癌细胞hct116和sw480)等方面具有显著活性。附图说明图1所示为本发明烯烃硫醚类订书肽的分子结构示意图。具体实施方式现有文献报道的制备订书肽的方法均是从氨基酸出发,首先进行保护,然后环化、选择性引入烯烃侧链,然后再开环,去掉保护基等,一般需要七八步反应才能制备得到。因其合成路线长、原料昂贵限制了其产品的应用。此外,早期订书肽中的线性多肽是将蛋白中的一段有功能的多肽截取出来,然后利用这段多肽中已有的氨基酸选择订合位点,这样的话可选择的订合位点很受限制,不利于订书桥的形成。半胱氨酸中含有巯基官能团,该巯基在订书肽的合成中常用作订合位点,但现有方法均将线性多肽中的半胱氨酸通过化学方法直接订合,即:线性多肽中不同位置有两个半胱氨酸,每个半胱氨酸的巯基残基都暴露在外,通过与二溴代烷烃反应,烷烃和巯基可以形成硫醚,从而将两个巯基残基连接起来,形成订书桥。用这种方法合成订书肽时,半胱氨酸上的巯基残基可能被包裹在线性多肽深部,使得反应困难。可见,该方法选择性低、收率不高、实用性差。针对以上缺陷,发明人在合成肽链时,首先在半胱氨酸的巯基上进行改造,引入烯烃基团,烯烃基团的引入不会改变半胱氨酸的手性和构型;其次,在设计好的位置接入能够形成订书桥的两个砌块(即形成订书桥的两个连接点或订合位点),肽链合成完毕后,再通过特定反应将两个砌块的末端连接,形成订书桥,从而得到订书肽。本发明还提出了一种新结构的订书肽,该订书肽中具有新的订书桥,即:半胱氨酸先与卤代烯烃进行醚化反应,在半胱氨酸上引入烯烃基团,制备得到烯烃醚化的半胱氨酸,并将其作为订书肽中形成订书桥的关键砌块;在合成线性肽时,将两个烯烃醚化的半胱氨酸引入线性肽链的不同位置,最后通过grubbs催化剂催化烯烃发生复分解反应,形成订书桥,实现多肽的订合,得到的烯烃硫醚类订书肽,如式(ⅰ)所示。本发明这种订合方法还未见报道。式(ⅰ)中,(x)n表示含有2-4和天然氨基酸的多肽片段,n为该多肽片段中氨基酸的数量,如2、3、4,(x)n中的n个氨基酸均为天然氨基酸,可相同,可不同,最好在w、k、l、f、d和s中选择;天然氨基酸有20种,分别为甘氨酸(g)、丙氨酸(a)、缬氨酸(v)、亮氨酸(l)、异亮氨酸(i)、苯丙氨酸(f)、脯氨酸(p)、色氨酸(w)、丝氨酸(s)、酪氨酸(y)、半胱氨酸(c)、蛋氨酸(m)、天冬酰胺(n)、谷氨酰胺(q)、苏氨酸(t)、天冬氨酸(d)、谷氨酸(e)、赖氨酸(k)、精氨酸(r)、组氨酸(h);m1和m2分别为1-3的整数(如1、2或3),y1为由前述20种天然氨基酸组成的多肽片段,多肽片段中氨基酸数量为3-8(如3、4、5、6、7或8);y2为由前述20种天然氨基酸中的任意一种,或这20种天然氨基酸组成的多肽片段,多肽片段中氨基酸数量为2-5(如2、3、4或5)。优选的,当y1为三肽、四肽或五肽时,y2为二肽、三肽、四肽或五肽;如,y1是ho-l-k-l-nh2的序列结构时,y2是ho-l-q-nh2的序列结构。当y1为六肽、七肽或八肽时,y2为二肽、三肽或四肽或为20种天然氨基酸中的任意一种;如,y1是ho-q-p-t-f-s-d-nh2的序列结构时,y2是ho-l-p-nh2的序列结构,或20种天然氨基酸中的任意一种。本发明使用烯烃醚化的半胱氨酸作为合成订书肽的关键砌块,烯烃醚化的半胱氨酸容易大量合成和制备且价格便宜。最后通过grubbs催化剂催化烯烃发生复分解反应,形成订书桥,该反应收率较高,且产生的杂质较少。制备式(ⅰ)烯烃硫醚类订书肽的方法,具体包括以下步骤:(1)半胱氨酸在碱的作用下与卤代烯烃(c3-c5)发生硫醚化反应,再在半胱氨酸的氨基端连接fmoc以保护氨基,得到烯烃醚化的非天然氨基酸fmoc-cys(c3-c5-ene)-oh,作为订书肽环合的关键砌块(即订书肽中形成订书桥的砌块);(2)使用wang树脂通过标准固相多肽合成方法合成得到线性多肽ho-y1-烯烃醚化的c-(x)n-烯烃醚化的c-y2-nh2,完成线性肽链的组装,其中烯烃醚化的c是步骤(1)得到的烯烃醚化的非天然氨基酸fmoc-cys(c3-c5-ene)-oh;(3)线性肽链组装完毕后,在反应体系中加入二氯甲烷和grubbs催化剂,线性肽链中两个烯烃醚化的非天然氨基酸fmoc-cys(c3-c5-ene)-oh的烯烃侧链在grubbs催化剂的催化下通过烯烃复分解反应连接起来,形成环的结构(即进行环合),当作为原料的线性肽链反应完毕后,反应结束;反应结束后使用三氟乙酸裂解液将订书肽粗品从树脂上切割下来,得到订书肽粗品;高压制备液相色谱分离纯化,冷冻干燥得到烯烃硫醚类订书肽。以下结合具体实施例,更具体地说明本发明的内容,并对本发明作进一步阐述,但这些实施例绝非对本发明进行限制。以下具体实施例说明本发明化合物的制备及其性质,实施例并非穷举。实施例中使用的试剂均为市场购买,核磁共振谱用日本电子jnm-eca-400超导核磁仪(400mhz)测定,esi质谱用美国安捷伦1260-g6230a型质谱仪测定,高分辨率质谱用美国布鲁克9.4t超高性能混合型串联傅里叶变换离子回旋共振质谱q-ft-icr-ms测定,maldi-tof质谱用德国brukerultraflexⅲmaldi-tof型质谱仪测定。实施例1:(xl-1)的合成(1)烯烃醚化的非天然氨基酸fmoc-cys(c3-c5-ene)-oh的合成fmoc-cys(allyl)-oh的合成l-半胱氨酸2.98g(24.7mmol)置于250ml烧瓶中,加入75ml乙醇,冰浴,将氢氧化钠3.50g(87.5mmol)溶于45ml水中后加入反应体系。室温搅拌10分钟,加入3-溴丙烯3.30g(24.7mmol),继续搅拌反应3小时。加入冰醋酸至反应体系ph=5.5,大量的白色固体析出,过滤,真空干燥,得到2.50g固体。将此固体置于100ml单口烧瓶中,加入50ml水,将1.90g碳酸氢钠(22.6mmol)分批加入反应体系内,6.54g9-芴甲基琥珀酰亚氨基碳酸酯(fmoc-osu)(19.4mmol)溶于25ml四氢呋喃中后,加入反应体系内,室温反应2小时。减压浓缩,水相调节ph=5,以150ml乙酸乙酯萃取水相,有机相以无水硫酸钠干燥,过滤,减压蒸出溶剂后经硅胶柱层析(石油醚:乙酸乙酯=1:1)得到2.50g淡黄色固体。1hnmr(400mhz,cdcl3)δ(ppm):12.88(1h,s),7.90(2h,d,j=7.6hz),7.74(2h,d,j=7.3hz),7.43(2h,t,j=7.4hz),7.33(2h,t,j=7.4hz),5.80(1h,m),5.07(1h,dd,j=1.7hz,j=17.1hz),5.00(1h,d,j=10.4hz),4.34-4.22(3h,m),4.14(1h,m),2.93(1h,dd,j=4.5hz,j=13.7hz),2.76(1h,dd,j=9.5hz,j=13.7hz),2.60(2h,m),2.27(2h,m)。esim/z:[m+h]+384.13,[m+na]+406.11,确证该淡黄色固体为fmoc-cys(allyl)-oh。fmoc-cys(butyl)-oh的合成将0.56gl半胱氨酸(4.6mmol)置于250ml单口烧瓶中,加入75ml乙醇,冰浴,将0.82g氢氧化钠(20.5mmol)溶于15ml水中后加入反应体系。室温搅拌10min,慢慢加入0.62g4-溴丁烯(4.6mmol),继续搅拌3h。加入冰醋酸至反应体系至ph=5.5,大量的白色固体析出,过滤,真空干燥,得到0.80g白色固体。将此固体置于50ml单口烧瓶中,加入10ml水,将0.67g碳酸氢钠(8.0mmol)分批加入反应体系内,1.51gfmoc-osu(4.5mmol)溶于6.0ml四氢呋喃后加入反应体系内,室温反应2小时。减压浓缩,调节ph=5,以150ml乙酸乙酯萃取水相,有机相以无水硫酸钠干燥,过滤,减压蒸出溶剂后经硅胶柱层析(石油醚:乙酸乙酯=1:1)得到0.40g淡黄色固体。1hnmr(400mhz,cdcl3)δ(ppm):7.89(2h,d,j=7.6hz),7.72(2h,d,j=7.3hz),7.41(2h,m),7.32(2h,m),6.94(1h,s),5.72(1h,m),5.09(1h,d,j=15.4hz),5.03(1h,d,j=9.8hz),4.30(1h,d,j=12.0hz),4.23(2h,m),3.91(1h,s),3.11(2h,d,j=7.3hz),2.91(1h,m),2.71(1h,m)。esim/z:[m+h]+398.13,[m+na]+420.11,[m+k]+436.08,确证该淡黄色固体为fmoc-cys(butyl)-oh。(2)固相合成ho-lklc(butyl)kfkc(allyl)lq-nh2序列称取0.30gwang树脂(负载率0.63mmol/g,0.19mmol)与201mgfmoc-leu-oh(n-芴甲氧羰基-l-亮氨酸,0.57mmol)混合于3ml二氯甲烷(dcm)和2滴n,n-二甲基甲酰胺(dmf)的混合溶剂中,加入4-二甲氨基吡啶(dmap)69mg(0.57mmol)搅拌使其充分溶解,冰浴下缓缓加入二异丙基碳二亚胺(dic)71mg(0.57mmol),反应于冰浴下搅拌30min后于室温搅拌24h,过滤,以1.5mldmf、1.5mldcm和1.5ml甲醇(meoh)依次洗涤后真空干燥,得到0.54g树脂。将树脂混悬于4ml吡啶-醋酸酐-dmf(v/v=1:1:2)混合溶液于固相合成管里,振摇1h后滤除反应液,树脂以1.5mldmf洗3次后用于后续反应。将树脂混悬于3ml20%(v/v)哌啶/dmf溶液,振摇15min后滤除反应液,树脂以1.5mldmf洗涤后,依次向树脂中加入267mgfmoc-lys(tbu)-oh(n-芴甲氧羰基-n’-叔丁氧羰基-l-赖氨酸,0.57mmol)、182mgo-苯并三氮唑-n,n,n’,n’-四甲基脲四氟硼酸(tbtu)(0.57mmol)和77mg1-羟基苯并三唑(hobt)(0.57mmol),再加入3mldmf于固相合成管中(其中有树脂),向体系缓缓滴加57mgn-甲基吗啡啉(nmm)(0.57mmol),反应振摇45-60min,过滤,树脂依次以1.5mldmf、1.5mldcm和1.5ml甲醇洗涤后真空干燥。按照缩合步骤逐一连接各氨基酸后脱除保护。(3)订书肽xl-1的合成将负载线性多肽的树脂0.3g与一代grubbs催化剂(苯亚甲基双(三环己基磷)二氯化钌)8.2mg(0.01mmol)溶解于6mldcm中,反应振摇24h。过滤,依次以4.5mldmf、4.5mldcm和4.5ml甲醇洗涤后真空干燥。加入3ml裂解液,裂解液为三氟乙酸(tfa):水(h2o):三异丙基硅烷(tis):间甲酚(m-cresol):1,2-乙二硫醇(1,2-ethanedithiol)=90:2.5:2.5:2.5:2.5(v/v),反应于室温下反应90min,过滤,滤液50℃减压浓缩,加入乙醚,离心3min,弃去上清液,沉淀以氩气吹干,沉淀即为粗品订书肽。将180mg粗品订书肽以乙腈和水的混合溶液溶解(水中含0.1%(v/v)三氟乙酸(tfa),乙腈和水的体积比为1:1),以微孔滤膜(nylon,0.45μm)过滤。样品溶液用装备c18制备柱的高效液相色谱仪进行分离纯化,流动相为乙腈:水(0.1%tfa溶液)=35:65。冷冻干燥得到目标订书肽(命名为xl-1)42mg。hrmsm/z:for[m]+,calculated1289.641,found1289.604.使用与xl-1相同的制备过程,仅将fmoc-cys(butyl)-oh变成fmoc-cys(allyl)-oh,得到式(2)化合物,且经确证为式(2)化合物。实施例2:(xl-2)的合成按实施例1的方法进行。0.30gwang树脂(负载率0.63mmol/g,0.19mmol)通过标准固相多肽合成方法进行合成。分离纯化后得到目标订书肽(命名为xl-2)38mg。hrmsm/z:for[m]+,calculated1289.641,found1289.604。使用与xl-2相同的制备过程,仅将fmoc-cys(butyl)-oh变成fmoc-cys(allyl)-oh,得到式(4)化合物,且经确证为式(4)化合物。实施例3:(xl-3)的合成按实施例1的方法进行。0.30gwang树脂(负载率0.63mmol/g,0.19mmol)通过标准固相多肽合成方法进行合成。分离纯化后得到目标订书(命名为xl-3)41mg。maldi-tofm/z:[m]+1476.0。使用与xl-3相同的制备过程,仅将一个fmoc-cys(allyl)-oh变成fmoc-cys(butyl)-oh,得到式(5)化合物,经确证为式(5)化合物。实施例4:(xl-4)的合成按实施例1的方法进行。0.30gwang树脂(负载率0.63mmol/g,0.19mmol)通过标准固相多肽合成方法进行合成。分离纯化后得到目标订书肽(命名为xl-4)36mg。hrmsm/z:for[m+na-h]+,calculated1498.664,found1498.830。使用与xl-4相同的制备过程,仅将一个fmoc-cys(allyl)-oh变成fmoc-cys(butyl)-oh,得到式(7)化合物,且经确证为式(7)化合物。实施例5:(xl-5)的合成按实施例1的方法进行。0.30gwang树脂(负载率0.63mmol/g,0.19mmol)通过标准固相多肽合成方法进行合成。分离纯化后得到目标订书肽(命名为xl-5)45mg。hrmsm/z:for[m]+,calculated1504.706,found1504.689。使用与xl-5相同的制备过程,仅将一个fmoc-cys(allyl)-oh变成fmoc-cys(butyl)-oh,得到式(10)化合物,且经确证为式(10)化合物。实施例6:(xl-6)的合成按实施例1的方法进行。0.30gwang树脂(负载率0.63mmol/g,0.19mmol)通过标准固相多肽合成方法进行合成。分离纯化后得到目标订书肽(命名为xl-6)39mg。hrmsm/z:for[m]+,calculated1504.706,found1504.691。使用与xl-6相同的制备过程,仅将一个fmoc-cys(allyl)-oh变成fmoc-cys(butyl)-oh,得到式(12)化合物,且经确证为式(12)化合物。实验一:抑制人乳腺癌细胞mcf-7增殖实验培养基及培养条件:dmem培养基:胎牛血清(v/v)=9:1,同时加入1%(v/v)的双抗(即终浓度为100u/ml青霉素和100u/ml的链霉素),37℃,5%co2环境下培养,得到培养基a。药物溶液配制:精密称定冻干的实施例1-6的订书肽适量,以dmem完全培养基为稀释液,配制浓度为500μmol/l的含药溶液,经0.22μm微孔滤膜过滤除菌,在无菌条件下分别将上述溶液用培养基a稀释至浓度分别为500、250、125、50、10μmol/l的订书肽溶液,现用现配。mcf-7细胞mtt实验:取对数生长期的mcf-7细胞,胰蛋白酶消化离心后,加入培养基a混悬细胞,血球计数板计数调节细胞浓度为1×105/ml,接种于96孔板,每孔100μl,周边一圈孔板中加入100μl的pbs(ph=7.4)保证细胞生长情况的均一。将上述96孔板放入培养箱中37℃,5%co2环境培养过夜,弃去旧培养基,将上述配制好的500、250、125、50、10μmol/l的订书肽溶液分别以每孔100μl的体积加入孔中,每个浓度设4个复孔。加药完毕放回培养箱中继续培养24h后,每孔加入20μlmtt(0.5mg/ml),37℃条件孵育4h,孵育完毕后吸弃上清液,每孔加入150μl的dmso,将板置于摇床振荡10min,酶标仪于490nm处测定各孔的od值。每个96孔板均设置对照孔及空白孔,细胞存活率用各孔的od值比值表示:细胞抑制率(%)=(od对照-od实验)/(od对照-od空白)×100%。以实施例1和2为例,实验结果见表1,表1中xl-1u和xl-2u分别为与xl-1和xl-2序列相同但未经环合形成订书桥的线性多肽。同时还以结构式为式a的化合物(l-029)作为阳性对照,l-029是具有抗肿瘤活性的小分子化合物,其制备方法及用途见公开号为cn103183665a的专利申请,其浓度为100、50、25、1、0.1μmol/l时,细胞抑制率分别为25.1%、8.5%、6.1%、5.0%、0,ic50为>100μm。表1订书肽xl-1和xl-2对mcf-7细胞的抑制作用xl-1xl-1uxl-2xl-2u500μm63.7%62.0%81.6%66.6%250μm64.0%55.4%83.2%64.9%125μm56.2%51.7%76.1%50.5%50μm54.7%25.7%75.9%41.2%10μm17.8%17.8%67.4%23.6%ic50(μm)44.6120<10.0119此实验表明,本发明的订书肽对mcf-7细胞有显著的抑制作用。此外,与没有订书桥但序列相同的线性多肽相比,采用本发明订合位点形成的订书肽对mcf-7细胞的抑制作用显著提高;并且跟阳性对照(l-029)相比,本发明的订书肽对mcf-7细胞的抑制作用也显著提高,说明本发明订书肽在抗乳腺癌方面具有较好的应用前景。实验二:抑制人结肠癌细胞hct116增殖实验培养基及培养条件:同实验一。药物溶液配制:同实验一。hct116细胞mtt实验:同实验一,仅是将实验一的mcf-7细胞换成hct116细胞。以实施例3和4为例,实验结果见表2,表2中xl-3u为与xl-3序列相似但未经环合形成订书桥的线性多肽,xl-3u的序列为ho-qptfsdlwkllp-nh2;xl-4u为与xl-4序列相同但未经环合形成订书桥的线性多肽。同时还以结构式为式a的化合物(l-029)作为阳性对照,得出其ic50为96μm。表2订书肽xl-3和xl-4对hct116细胞的抑制作用此实验表明,本发明的订书肽对hct116细胞有显著的抑制作用。此外,与没有订书桥的序列相同或相似的线性多肽相比,采用本发明订合位点形成的订书肽对hct116细胞的抑制作用显著提高;并且跟阳性对照(l-029)相比,本发明的订书肽对hct116细胞的抑制作用也显著提高,说明本发明订书肽在抗结肠癌方面具有较好的应用前景。实验三:抑制人结肠癌细胞sw480增殖实验培养基及培养条件:同实验一。药物溶液配制:同实验一。sw480细胞mtt实验:同实验一,仅是将实验一的mcf-7细胞换成sw480细胞。以实施例3和4为例,实验结果见表3,表3中xl-3u为与xl-3序列相似但未经环合形成订书桥的线性多肽,xl-3u的序列为ho-qptfsdlwkllp-nh2;xl-4u为与xl-4序列相同但未经环合形成订书桥的线性多肽。同时还以结构式为式a的化合物(l-029)作为阳性对照,得出其ic50为76μm。表3订书肽xl-3和xl-4对sw480细胞的抑制作用xl-3xl-3uxl-4xl-4u500μm80.0%58.9%88.4%41.8%250μm77.9%56.4%86.0%15.7%125μm72.3%45.7%72.5%15.0%50μm68.6%45.6%61.3%14.3%10μm66.9%19.2%95.1%17.2%ic50(μm)<10.017243>500此实验表明,本发明的订书肽对sw480细胞有显著的抑制作用。此外,与没有订书桥的序列相同或相似的线性多肽相比,采用本发明订合位点形成的订书肽对sw480细胞的抑制作用显著提高;并且跟阳性对照(l-029)相比,本发明的订书肽对sw480细胞的抑制作用也显著提高,说明本发明订书肽在抗结肠癌方面具有较好的应用前景。以上所述仅是本发明的优选实施方式,应当指出的是,对于本
技术领域:
的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的内容。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1