含碳物质的材料的制造方法、含碳物质的材料、和可溶性有机无机复合体与流程
1.本发明涉及含碳物质的材料的制造方法、含碳物质的材料和可溶性有机无机复合体。
背景技术:
2.考虑到近来资源节约和能源节约的趋势,已经开发了轻质碳材料,并且将其用于各种领域。例如,已经报道了含碳物质的材料,例如碳被覆无机颗粒和中空碳细颗粒作为各自包括这种轻质碳材料并且可用于各种领域的化合物(例如,专利文献1~4,和非专利文献1~4)。另外,已知活性炭、炭黑等为工业上制造的含碳物质的材料。
3.然而,难以将诸如碳被覆无机颗粒和中空碳细颗粒等的现有技术的含碳物质的材料应用于以低成本进行批量生产的工业生产中,这是因为该材料需要通过在要被覆有碳的各无机颗粒和各自用作中空结构的基料的细颗粒的表面上例如借由气相反应或高温沉积反应形成碳物质膜来制造。
4.另外,如上所述的工业地制造的包括活性炭、炭黑等的含碳物质的材料具有各种官能团。因此,存在的问题在于,难以精确地控制含碳物质的材料的结构,并且因此发生其物性的变化。近年来,需要能够肯定地表现出目标物性的含碳物质的材料,因此需要开发具有精确控制的结构的含碳物质的材料。
5.引文列表
6.专利文献
7.[ptl 1]jp 07
‑
187849 a
[0008]
[ptl 2]jp 07
‑
267618 a
[0009]
[ptl 3]jp 2005
‑
281065 a
[0010]
[ptl 4]jp 2010
‑
168251 a
[0011]
非专利文献
[0012]
[npl 1]dawei pan et.al.,langmuir,22,5872
‑
5876(2006)
[0013]
[npl 2]v.ruiz et.al.,electrochemistry communications,24,35
‑
38(2012)
[0014]
[npl 3]h.nishihara et.al.,adv.funct.mater.,26,6418
‑
6427(2016)
[0015]
[npl 4]riichiro saito,"advanced technologies and emerging applications of graphene",chapter 2.basic physical properties of graphene and chapter 3.photoelectronic physical properties of graphene
技术实现要素:
[0016]
发明要解决的问题
[0017]
本发明的目的是提供一种在温和条件下简单地制造在溶剂中具有溶解性的含碳物质的材料或具有精确控制的结构的含碳物质的材料的方法。本发明的另一个目的是提供
一种含碳物质的材料,其中碳物质和无机物通过共价键彼此地结合。本发明的又一个目的是提供一种有机无机复合体,其可用于例如工业上制造含碳物质的材料,例如碳被覆无机颗粒和中空碳细颗粒,并且可以在温和条件下工业生产。
[0018]
用于解决问题的方案
[0019]
根据本发明的一个实施方案,提供一种含碳物质的材料的制造方法,其包括加热含有化合物(a)和无机物的组合物的加热步骤(i),所述化合物(a)通过加热而引起相同分子之间和/或不同分子之间的缩合反应,其中当所述化合物(a)的缩合反应温度为t℃时,所述加热步骤(i)中的加热温度为(t
‑
150)℃以上,
[0020]
条件是本文中使用的缩合反应温度t是指在不存在催化剂(载体)时的缩合反应温度。
[0021]
在一个实施方案中,含碳物质的材料的制造方法还包括在所述加热步骤(i)之后将通过加热所述化合物(a)制造的碳物质的至少一部分除去的碳物质除去步骤。
[0022]
在一个实施方案中,含碳物质的材料的制造方法还包括在所述碳物质除去步骤之后进一步加热残留物的加热步骤(ii)。
[0023]
在一个实施方案中,含碳物质的材料的制造方法还包括在所述加热步骤(i)之后将所述无机物除去的无机物除去步骤。
[0024]
在一个实施方案中,含碳物质的材料的制造方法还包括在所述无机物除去步骤之后进一步加热残留物的加热步骤(ii)。
[0025]
在一个实施方案中,所述化合物(a)的分子量为500以下。
[0026]
在一个实施方案中,所述化合物(a)的缩合反应温度为450℃以下。
[0027]
在一个实施方案中,所述化合物(a)的缩合反应温度为400℃以下。
[0028]
在一个实施方案中,当将所述化合物(a)在氮气气氛下、从40℃起在10℃/min的升温条件下进行tg
‑
dta分析时,其在温度500℃下的重量m500与其在温度50℃下的初始重量m50的重量比(m500/m50)为0.2以上。
[0029]
在一个实施方案中,所述缩合反应通过酸催化剂来促进。
[0030]
在一个实施方案中,所述缩合反应为选自由以下组成的组中的至少一种:
[0031]
(a)由
‑
h基与
‑
oh基形成h2o并脱离而发生的缩合反应;
[0032]
(b)由
‑
h基与
‑
or基形成roh并脱离而发生的缩合反应,其中r表示任意适当的取代或未取代的烷基;
[0033]
(c)由
‑
h基与
‑
x基形成hx并脱离而发生的缩合反应,其中x表示卤素或cn;
[0034]
(d)由
‑
h基与
‑
nh2基形成nh3并脱离而发生的缩合反应;
[0035]
(e)由
‑
h基与
‑
nhr基形成rnh2并脱离而发生的缩合反应,其中r表示任意适当的取代或未取代的烷基;
[0036]
(f)由
‑
h基与
‑
nr1r2基形成r1r2nh并脱离而发生的缩合反应,其中r1和r2各自表示任意适当的取代或未取代的烷基;
[0037]
(g)由
‑
h基与
‑
sh基形成h2s并脱离而发生的缩合反应;
[0038]
(h)由
‑
h基与
‑
sr基形成rsh并脱离而发生的缩合反应,其中r表示任意适当的取代或未取代的烷基;
[0039]
(i)由
‑
h基与
‑
oocr基形成rcooh并脱离而发生的缩合反应,其中r表示任意适当的
取代或未取代的烷基;
[0040]
(j)由
‑
h基与
‑
oso(oh)基形成h2so3并脱离而发生的缩合反应;
[0041]
(k)由
‑
h基与
‑
oso2r基形成rso2(oh)并脱离而发生的缩合反应,其中r表示任意适当的取代或未取代的烷基;
[0042]
(l)由
‑
h基与
‑
oso2(or)基形成roso3h并脱离而发生的缩合反应,其中r表示任意适当的取代或未取代的烷基;和
[0043]
(m)由
‑
h基与
‑
oso2(oh)基形成h2so4并脱离而发生的缩合反应。
[0044]
在一个实施方案中,所述无机物为选自由无机氧化物、无机氮化物、无机硫化物、无机碳化物和不溶性盐组成的组中的至少一种。
[0045]
在一个实施方案中,所述无机氧化物为各自在其表面上具有官能团的无机氧化物颗粒。
[0046]
在一个实施方案中,所述无机氧化物颗粒为选自由二氧化硅颗粒、氧化铝颗粒、二氧化钛颗粒、氧化镁颗粒、多酸颗粒、其表面被至少部分地氧化的金属颗粒、复合氧化物颗粒和固溶体氧化物颗粒组成的组中的至少一种。
[0047]
在一个实施方案中,用于形成所述多酸颗粒的金属为选自由钼、钒、钨、铌、钛和钽组成的组中的至少一种。
[0048]
在一个实施方案中,所述无机氧化物的分解温度为800℃以上。
[0049]
根据本发明的一个实施方案,提供一种含碳物质的材料,其包括:
[0050]
碳物质;和
[0051]
无机物,
[0052]
其中所述碳物质的至少一部分和所述无机物的至少一部分通过共价键结合在一起。
[0053]
在一个实施方案中,本发明的含碳物质的材料在其
13
c
‑
nmr分析中在125ppm~135ppm范围内显示峰。
[0054]
在一个实施方案中,本发明的含碳物质的材料在其
13
c
‑
nmr分析中在140ppm~160ppm范围内显示峰。
[0055]
根据本发明的一个实施方案,提供一种有机无机复合体,其包括:
[0056]
碳物质;和
[0057]
无机物,
[0058]
其中所述碳物质为溶剂可溶的。
[0059]
在一个实施方案中,所述溶剂为n
‑
甲基吡咯烷酮。
[0060]
在一个实施方案中,所述无机物为选自由无机氧化物、无机氮化物、无机硫化物、无机碳化物和不溶性盐组成的组中的至少一种。
[0061]
在一个实施方案中,所述无机氧化物为各自在其表面上具有官能团的无机氧化物颗粒。
[0062]
在一个实施方案中,所述无机氧化物颗粒为选自由二氧化硅颗粒、氧化铝颗粒、二氧化钛颗粒、氧化镁颗粒、多酸颗粒、其表面被至少部分地氧化的金属颗粒、复合氧化物颗粒和固溶体氧化物颗粒组成的组中的至少一种。
[0063]
在一个实施方案中,用于形成所述多酸颗粒的金属为选自由钼、钒、钨、铌、钛和钽
组成的组中的至少一种。
[0064]
发明的效果
[0065]
根据本发明,可以提供一种在温和条件下简单地制造在溶剂中具有溶解性的含碳物质的材料或具有精确控制的结构的含碳物质的材料的方法。根据本发明,还可以提供其中碳物质和无机物通过共价键彼此结合的含碳物质的材料。根据本发明,还可以提供有机
‑
无机复合物,其可用于例如工业生产的含碳物质的材料,例如碳被覆无机颗粒和中空碳细颗粒,并且可以在温和条件下工业地制造。
附图说明
[0066]
图1是用于说明作为通过本发明的制造方法得到的含碳物质的材料的一实施方案的有机无机复合体以及本发明的有机无机复合体各自的一个优选实施方案的示意性截面图。
[0067]
图2是用于说明作为通过本发明的制造方法得到的含碳物质的材料的一个实施方案的含碳物质的颗粒的一例的核壳颗粒和可以从本发明的有机
‑
无机复合体得到的核壳颗粒的示意性截面图。
[0068]
图3是实施例1~4中得到的各有机无机复合体(1)~(4)的xps光谱(c1s)图。
[0069]
图4是用于显示实施例1~4中得到的各有机无机复合体(1)~(4)的tg
‑
dta分析中的dta分析结果的测量图。
[0070]
图5是实施例1~4中得到的各有机无机复合体(1)~(4)的ir光谱图。
[0071]
图6是实施例1~4中得到的各有机无机复合体(1)~(4)的拉曼光谱图。
[0072]
图7是实施例5~8中得到的各有机无机复合体(5)~(8)的xps光谱(c1s)图。
[0073]
图8是实施例5~8中得到的各有机无机复合体(5)~(8)的ir光谱图。
[0074]
图9是实施例9中得到的有机无机复合体(9)的拉曼光谱图。
[0075]
图10是实施例9中得到的高度碳化的核壳颗粒(9)的sem照片图。
[0076]
图11是实施例9中得到的高度碳化的核壳颗粒(9)的表面的拉曼光谱图。
[0077]
图12是实施例9中得到的高度碳化的核壳颗粒(9)和比较例3中得到的颗粒各自的
13
c
‑
nmr图。
[0078]
图13是实施例9中得到的高度碳化的核壳颗粒(9)、比较例3中得到的颗粒和二氧化硅颗粒各自的
29
si
‑
nmr图。
[0079]
图14是实施例10中得到的高度碳化的核壳颗粒(10)的表面的拉曼光谱图。
[0080]
图15是实施例11中得到的高度碳化的核壳颗粒(11)的表面的拉曼光谱图。
[0081]
图16是用于比较通过高度碳化的核壳颗粒(11)的sps烧结得到的烧结体与通过铝的sps烧结得到的烧结体的维氏硬度的图形表现。
[0082]
图17是实施例12中得到的高度碳化的核壳颗粒(12)的表面的拉曼光谱图。
[0083]
图18是实施例13中得到的高度碳化的核壳颗粒(13)的表面的拉曼光谱图。
[0084]
图19是实施例14中得到的高度碳化的核壳颗粒(14)的表面的拉曼光谱图。
[0085]
图20是实施例15中得到的高度碳化的核壳颗粒(15)的表面的拉曼光谱图。
[0086]
图21是实施例16中得到的高度碳化的核壳颗粒(16)的表面的拉曼光谱图。
[0087]
图22是实施例17中得到的高度碳化的核壳颗粒(17)的表面的拉曼光谱图。
[0088]
图23是实施例18中得到的高度碳化的核壳颗粒(18)的表面的拉曼光谱图。
[0089]
图24是实施例19中得到的高度碳化的核壳颗粒(19)的表面的拉曼光谱图。
[0090]
图25是实施例20中得到的高度碳化的核壳颗粒(20)的表面的拉曼光谱图。
[0091]
图26是实施例21中得到的有机无机复合体(21)的表面的拉曼光谱图。
[0092]
图27是实施例21中得到的高度碳化的核壳颗粒(21)的表面的拉曼光谱图。
[0093]
图28是实施例22中得到的有机无机复合体(22)的表面的拉曼光谱图。
[0094]
图29是实施例22中得到的高度碳化的核壳颗粒(22)的表面的拉曼光谱图。
[0095]
图30是实施例23中得到的有机无机复合体(23)的表面的拉曼光谱图。
[0096]
图31是实施例23中得到的高度碳化的核壳颗粒(23)的表面的拉曼光谱图。
[0097]
图32是实施例24中得到的有机无机复合体(24)的表面的拉曼光谱图。
[0098]
图33是实施例24中得到的高度碳化的核壳颗粒(24)的表面的拉曼光谱图。
具体实施方式
[0099]
<<<<1.含碳物质的材料的制造方法>>>>
[0100]
本发明的含碳物质的材料的制造方法包括加热含有化合物(a)和无机物的组合物的加热步骤(i),所述化合物(a)通过加热而引起相同分子之间和/或不同分子之间的缩合反应,其中当化合物(a)的缩合反应温度为t℃时,加热步骤(i)中的加热温度为(t
‑
150)℃以上。
[0101]
在加热步骤(i)中,加热含有化合物(a)和无机物的组合物,该化合物(a)通过加热而引起相同分子之间和/或不同分子之间的缩合反应。化合物(a)与无机物的共混比例如下:化合物(a)的量优选为0.01wt%~1,000,000wt%,更优选0.1wt%~100,000wt%,特别优选1wt%~1,000wt%,相对于100wt%的无机物。当化合物(a)与无机物的共混比例落在上述范围时,可以在更温和的条件下更简单地制造具有更精确控制的结构的含碳物质的材料。无机物与化合物(a)之间的共混比例可以根据复合体的目标物性适当调节。例如,调节无机物与化合物(a)之间的共混比例能够控制要获得的含碳物质的材料的物性和形态(例如,在溶剂中的溶解度,碳组分或无机组分的形状(不论该组分是颗粒状或非颗粒状),以及碳组分或无机组分的尺寸)。
[0102]
可以在本发明的效果不受损的程度将任何适当的其他组分掺入含有化合物(a)和无机物的组合物中,该化合物(a)通过加热而引起相同分子之间和/或不同分子之间的缩合反应。这种其他组分的实例包括溶剂、催化剂、母体材料和载体。
[0103]
在加热步骤(i)中待加热的组合物在本发明的效果不受损的程度仅需要通过任何适当的方法来制备即可。这种方法例如是包括当组分为固态时通过任何适当的方法(例如,压碎或粉碎)将化合物(a)和无机物混合的方法。另外,该方法例如是包括通过任何适当的方法(例如,超声波处理)将化合物(a)、无机物、溶剂以及根据需要的除溶剂之外的任何其他组分混合并通过任何适当的方法(例如真空干燥)除去溶剂的方法。另外,可以根据需要进行切碎。
[0104]
当化合物(a)的缩合反应温度为t℃时,加热步骤(i)中的加热温度为(t
‑
150)℃以上,优选(t
‑
150)℃~(t+50)℃,更优选(t
‑
130)℃~(t+45)℃,还更优选(t
‑
100)℃~(t+40)℃,特别优选(t
‑
80)℃~(t+35)℃,最优选(t
‑
50)℃~(t+30)℃。在本发明的含碳物质
的材料的制造方法中,无机物的催化能力以及无机物上的官能团与碳物质之间的反应性高,因此官能团与碳物质之间的反应可以从与如上所述的化合物(a)的缩合反应温度相比为相对较低的温度进行,以推进无机物的碳化。当将加热温度调节在上述范围时,可以在更温和的条件下更简单地制造在溶剂中具有溶解性的含碳物质的材料或具有更精确控制的结构的含碳物质的材料。
[0105]
化合物(a)的缩合反应温度可以通过tg
‑
dta分析确定。具体程序如下所述。
[0106]
(1)当使用一种化合物作为化合物(a)时,在氮气气氛下、且从40℃起以10℃/min的升温速度进行化合物(a)的tg
‑
dta分析,将所得的dta曲线的最低峰顶温度确定为化合物(a)的缩合反应温度(t℃)。
[0107]
(2)当使用两种以上的化合物的混合物作为化合物(a)时,在氮气气氛下、且从40℃起以10℃/min的升温速度进行混合物的tg
‑
dta分析,将所得的dta曲线的最低峰顶温度确定为化合物(a)(两种以上的化合物的混合物)的缩合反应温度(t℃)。
[0108]
(3)但是,当作为一种化合物或两种以上的化合物的混合物的化合物(a)包含例如杂质如溶剂、水分或水合水时,在某些情况下,在低于缩合反应温度的温度下观察到伴随杂质的脱离的dta峰(有时称为“杂质峰”)。在这种情况下,在忽略杂质峰的同时,确定化合物(a)的缩合反应温度。在忽略杂质峰的同时,典型地将所得的dta曲线的最低峰顶温度确定为化合物(a)的缩合反应温度。
[0109]
作为具体的加热温度,在加热步骤(i)中的加热温度优选为200℃~500℃,更优选220℃~400℃,还更优选230℃~350℃,最优选250℃~300℃。当将加热温度调节在上述范围时,可以在更温和的条件下更简单地制造在溶剂中具有溶解性的含碳物质的材料或具有更精确控制的结构的含碳物质的材料。特别地,如上所述加热步骤(i)中的加热温度低,因此含碳物质的材料可以在更温和的条件下工业地制造。
[0110]
作为具体的加热时间,在加热步骤(i)中的加热时间优选为0.1小时~120小时,更优选0.5小时~100小时,还更优选1小时~50小时,最优选2小时~24小时。当将加热时间调节在上述范围时,可以在更温和的条件下更简单地制造在溶剂中具有溶解性的含碳物质的材料或具有更精确控制的结构的含碳物质的材料。
[0111]
<<1
‑
1.化合物(a)>>
[0112]
化合物(a)通过加热而引起相同分子之间和/或不同分子之间的缩合反应,因此化合物(a)可以典型地通过加热含有化合物(a)和无机物的组合物的加热步骤(i)而转变成碳物质。
[0113]
优选的是,化合物(a)在23℃的环境下为固体且具有熔点。当该化合物具有熔点时,该化合物在其煅烧过程中熔融,因此分子之间的反应令人满意地进行。如果该化合物不具有任何熔点,则该化合物在煅烧过程中不熔融。因此,分子的位置是固定的,并由此几乎不促进分子之间的反应。因而,该化合物难以转变成碳物质。当采用这样的化合物(a)时,缩合反应加速,并由此抑制了该化合物的分解反应,或者要得到的含碳物质的材料中的碳物质在溶剂中的溶解性变得更优异(例如,碳物质中溶解在溶剂中的组分的量变大,或者可溶解碳物质的溶剂的种类数增加)。
[0114]
对缩合没有贡献的化合物(a)的骨架优选为芳族结构。当骨架为芳族时,要得到的碳物质的碳组分可以更稳定。这种芳族结构优选例如为:包含碳原子的芳族结构,例如苯或
萘;或包含碳原子和杂原子(例如氮或氧)的杂芳族结构,例如吡啶、嘧啶、呋喃或噻吩。其中,更优选各自具有六元环结构的芳族结构和杂芳族结构,例如苯和吡啶。
[0115]
可以在本发明效果不受损的程度采用任何适当的分子量作为化合物(a)的分子量。这种分子量优选为500以下,更优选75~450,还更优选80~400,最优选100~350,因为可以进一步表现本发明的效果。
[0116]
可以在本发明效果不受损的程度采用任何适当的缩合反应温度作为化合物(a)的缩合反应温度。这种缩合反应温度优选为450℃以下,更优选400℃以下,还更优选200℃~370℃,特别优选250℃~350℃,因为可以进一步表现本发明的效果。
[0117]
当将化合物(a)在氮气气氛下、且从40℃起在10℃/min的升温条件下进行tg
‑
dta分析时,其在温度500℃下的重量m500与其在温度50℃下的初始重量m50的重量比(m500/m50)优选为0.2以上,更优选0.2~0.9,最优选0.3~0.8,因为可以进一步表现本发明的效果。当使用重量比(m500/m50)落在上述范围内的化合物(a)时,碳物质在加热后可以充分残留在含碳物质的材料中。
[0118]
[化合物(a)的典型实施方案(第一实施方案)]
[0119]
化合物(a)的典型实施方案(第一实施方案)为通过加热分解而在芳香环上产生自由基的芳族化合物。在芳香环上产生自由基的芳族化合物可以引起相同分子之间和/或不同分子之间的缩合反应,从而转变成碳物质。
[0120]
通过加热分解而在芳香环上产生自由基的芳族化合物优选为通过加热产生气体(在常温常压下处于气态的气体)的芳族化合物。
[0121]
可以采用任何适当的芳族化合物作为通过加热产生气体的芳族化合物,只要该化合物是芳族化合物并通过加热而产生气体即可。这种在常温常压下处于气态的气体优选为选自co、co2、n2、o2、h2和no2中的至少一种。
[0122]
当如上所述这种在常温常压下处于气态的气体是选自co、co2和o2中的至少一种时,要得到的含碳物质的材料优选具有特别是选自由以下方面(1)~(6)组成的组中的至少一种方面。
[0123]
(1)所有碳
‑
氧键,即c
‑
o键(包括醇衍生的c
‑
o键、醚衍生的c
‑
o键和环氧衍生的c
‑
o键)和c=o键(包括羰基衍生的c=o键、羧基衍生的c=o键、酯衍生的c=o键和内酯衍生的c=o键)的总量相对于所有键,即c
‑
c键、c=c键、c
‑
h键、c
‑
o键和c=o键的总量的比例(该比例由c1s xps分析确定)优选为10%以上,更优选20%以上,还更优选25%以上,并且该比例的上限优选为35%以下。当含碳物质的材料中c
‑
o键和c=o键的总量相对于c
‑
c键、c=c键、c
‑
h键、c
‑
o键和c=o键的总量的比例(该比例由c1s xps分析确定)落在上述范围时,含碳物质的材料不同于迄今已知的简单碳物质,可以是具有各种物性如溶解性的新型含碳物质的材料。
[0124]
(2)醚衍生的c
‑
o键(即,c
‑
o
‑
c键)和醇衍生的c
‑
o键(即,c
‑
oh键)的总量相对于所有碳
‑
氧键,即c
‑
o键(包括醇衍生的c
‑
o键、醚衍生的c
‑
o键和环氧衍生的c
‑
o键)和c=o键(包括羰基衍生的c=o键、羧基衍生的c=o键、酯衍生的c=o键和内酯衍生的c=o键)的总量的比例(该比例由c1s xps分析确定)优选为50%以上,更优选60%以上,还更优选65%以上,特别优选70%以上,最优选75%以上。该比例的上限优选为90%以下。当含碳物质的材料中醚衍生的c
‑
o键和醇衍生的c
‑
o键的总量相对于c
‑
o键和c=o键的总量的比例(该比例
由c1s xps分析确定)落在上述范围时,可以改进含碳物质的材料的碳物质部分的结构控制率,并且可以更精确地控制其结构。术语“结构控制率”是指源自于期望反应的键数与总键数的比例。本发明中,总键数对应于c
‑
o键和c=o键的总量,并且源自于期望反应的键数对应于醚衍生的c
‑
o键和醇衍生的c
‑
o键的总量。高的结构控制率换言之如下:源自于期望反应的键数大,而源自于不期望的反应的键数小。本发明中,源自于不期望的反应的键是源自碳物质的分解反应的c=o键。随着结构控制率变大,分解反应受到抑制,因此这种含碳物质的材料可以说是具有更精确控制的结构的含碳物质的材料。
[0125]
(3)醚衍生的c
‑
o键(即,c
‑
o
‑
c键)和醇衍生的c
‑
o键(即,c
‑
oh键)的总量相对于所有键,即c
‑
c键、c=c键、c
‑
h键、c
‑
o键(包括醇衍生的c
‑
o键、醚衍生的c
‑
o键和环氧衍生的c
‑
o键)和c=o键(包括羰基衍生的c=o键、羧基衍生的c=o键、酯衍生的c=o键和内酯衍生的c=o键)的总量的比例(该比例由c1s xps分析确定)优选为15%以上,更优选17%以上,还更优选20%以上。该比例的上限优选为30%以下。当含碳物质的材料中醚衍生的c
‑
o键(即,c
‑
o
‑
c键)和醇衍生的c
‑
o键(即,c
‑
oh键)的总量相对于所有键的总量的比例(该比例由c1s xps分析确定)落在上述范围时,含碳物质的材料可以说是具有更精确控制的结构的含碳物质的材料。
[0126]
(4)方面:其中所有碳
‑
氧键,即c
‑
o键(包括醇衍生的c
‑
o键、醚衍生的c
‑
o键和环氧衍生的c
‑
o键)和c=o键(包括羰基衍生的c=o键、羧基衍生的c=o键、酯衍生的c=o键和内酯衍生的c=o键)的总量相对于所有键,即c
‑
c键、c=c键、c
‑
h键、c
‑
o键和c=o键的总量的比例(该比例由c1s xps分析确定)落在方面(1)中所述的范围;以及其中醚衍生的c
‑
o键(即,c
‑
o
‑
c键)和醇衍生的c
‑
o键(即,c
‑
oh键)的总量相对于所有碳
‑
氧键,即c
‑
o键(包括醇衍生的c
‑
o键、醚衍生的c
‑
o键和环氧衍生的c
‑
o键)和c=o键(包括羰基衍生的c=o键、羧基衍生的c=o键、酯衍生的c=o键和内酯衍生的c=o键)的总量的比例(该比例由c1s xps分析确定)落在方面(2)中所述的范围。根据这样的方面,可以进一步改进含碳物质的材料的碳物质部分的溶解性,并且可以进一步改进碳物质部分的结构控制率。另外,根据这样的方面,可以甚至更精确地控制含碳物质的材料的结构。
[0127]
(5)含碳物质的材料在其ir分析中优选不显示由c=o拉伸振动导致的在1,660cm
‑1~1,800cm
‑1范围内的峰。当在含碳物质的材料的ir分析中在1,660cm
‑1~1,800cm
‑1范围内未观察到由c=o拉伸振动导致的峰时,可以更精确地控制含碳物质的材料的结构。当在含碳物质的材料的ir分析中在1,660cm
‑1~1,800cm
‑1范围内未观察到由c=o拉伸振动导致的峰时可以更精确地控制含碳物质的材料的结构的原因如上所述。
[0128]
(6)含碳物质的材料中的碳物质是溶剂可溶的。
[0129]
通过加热产生co和/或co2的芳族化合物的实例是具有
“‑
c(=o)
‑”
和/或
“‑
o
‑
c(=o)
‑”
结构的芳族化合物(例如芳族酮衍生物、芳族酯衍生物或酸酐)。
[0130]
通过加热产生co和/或co2的芳族化合物的实例包括以下化合物。
[0131][0132]
通过加热产生n2的芳族化合物的实例为具有"
‑
nh
‑
nh
‑
"结构、"
‑
n=n
‑
"结构或"
‑
n3"结构的芳族化合物(例如,芳族偶氮化合物、芳族叠氮化、三唑取代的芳族化合物、四唑取代的芳族化合物、三嗪或其衍生物、四嗪或其衍生物、或者芳族酰肼衍生物)。
[0133]
通过加热产生n2的芳族化合物的实例包括以下化合物。在以下化合物中,r表示氢原子,或者可具有取代基的烷基、芳基、或杂芳基。
[0134][0135]
通过加热产生o2的芳族化合物的实例为具有"
‑
o
‑
o
‑
"结构的芳族化合物(例如,芳族碳氧化物或芳族过氧化物)。
[0136]
通过加热产生o2的芳族化合物的实例包括以下化合物。在以下化合物中,r表示氢原子,或者可具有取代基的烷基、芳基、或杂芳基。
[0137]
[0138]
通过加热产生h2的芳族化合物的实例为具有"
‑
ch2‑
"结构的稠和多环芳族化合物(例如,非那烯系化合物)。
[0139]
通过加热产生h2的芳族化合物的实例包括以下化合物。
[0140][0141]
通过加热产生no2的芳族化合物的实例为具有"
‑
no2"结构的芳族化合物(例如,芳族硝基化合物)。
[0142]
通过加热产生no2的芳族化合物的实例为以下化合物。
[0143][0144]
通过加热分解而在芳香环上产生自由基的芳族化合物为以下化合物:该化合物具有热分解性,并且气体分子(优选为选自co、co2、n2、o2、h2和no2中的至少一种)是通过至少部分骨架的解离和分解产生的,而自由基则是在残留的芳香环上产生的。当使用这种芳族化合物时,反应仅由其自身的分解引起,而不需要任何反应催化剂。因此,可以抑制化学反应的副产物和反应催化剂存在于碳物质中而成为致命的杂质,并且由此可以得到质量更高的碳物质。另外,当使用这种芳族化合物时,可以在相对温和的温度环境下得到碳物质,而无需使用任何可燃性气体。另外,这样的芳族化合物可具有如此高的反应性以至于不需要催化作用。
[0145]
[化合物(a)的典型实施方案(第二实施方案)]
[0146]
化合物(a)的典型实施方案(第二实施方案)是一个中性分子由两种以上的基团通过它们的缩合反应形成并脱离这样的化合物。在第二实施方案中,允许一种化合物具有两种以上的基团的情况,并且也允许两种以上的化合物的各个基团组合以提供两种以上的基团的情况。这样的化合物(a)会导致相同分子之间和/或不同分子之间的缩合反应从而转变成碳物质。
[0147]
可以在本发明的效果不受损的程度采用任何适当的缩合反应作为缩合反应,只要该缩合反应是通过由两种以上的基团形成一个中性分子并脱离而发生的即可。当采用这种缩合反应时,该反应可以在相对低的温度下进行。这种缩合反应可以是,例如:
[0148]
(a)由
‑
h基与
‑
oh基形成h2o并脱离而发生的缩合反应;
[0149]
(b)由
‑
h基与
‑
or基形成roh并脱离而发生的缩合反应,其中r表示任意适当的取代或未取代的烷基;
[0150]
(c)由
‑
h基与
‑
x基形成hx并脱离而发生的缩合反应,其中x表示卤素或cn;
[0151]
(d)由
‑
h基与
‑
nh2基形成nh3并脱离而发生的缩合反应;
[0152]
(e)由
‑
h基与
‑
nhr基形成rnh2并脱离而发生的缩合反应,其中r表示任意适当的取代或未取代的烷基;
[0153]
(f)由
‑
h基与
‑
nr1r2基形成r1r2nh并脱离而发生的缩合反应,其中r1和r2各自表示任意适当的取代或未取代的烷基;
[0154]
(g)由
‑
h基与
‑
sh基形成h2s并脱离而发生的缩合反应;
[0155]
(h)由
‑
h基与
‑
sr基形成rsh并脱离而发生的缩合反应,其中r表示任意适当的取代或未取代的烷基;
[0156]
(i)由
‑
h基与
‑
oocr基形成rcooh并脱离而发生的缩合反应,其中r表示任意适当的取代或未取代的烷基;
[0157]
(j)由
‑
h基与
‑
oso(oh)基形成h2so3并脱离而发生的缩合反应;
[0158]
(k)由
‑
h基与
‑
oso2r基形成rso2(oh)并脱离而发生的缩合反应,其中r表示任意适当的取代或未取代的烷基;
[0159]
(l)由
‑
h基与
‑
oso2(or)基形成roso3h并脱离而发生的缩合反应,其中r表示任意适当的取代或未取代的烷基;和
[0160]
(m)由
‑
h基与
‑
oso2(oh)基形成h2so4并脱离而发生的缩合反应。
[0161]
特别地,当脱离的中性组分在脱离温度(煅烧温度)下为气体组分时,该组分存在于气相部分中而不被引入碳物质中,因此几乎不作为杂质。
[0162]
特别地,在上述缩合反应中的以下任何一种缩合反应的情况下,要获得的含碳物质的材料优选特别地具有选自由以下方面(1)~(6)组成的组中的至少一种方面:
[0163]
(a)由
‑
h基与
‑
oh基形成h2o并脱离而发生的缩合反应;
[0164]
(b)由
‑
h基与
‑
or基(其中r表示任意适当的取代或未取代的烷基)形成roh并脱离而发生的缩合反应;
[0165]
(i)由
‑
h基与
‑
oocr基(其中r表示任意适当的取代或未取代的烷基)形成rcooh并脱离而发生的缩合反应。
[0166]
(1)所有碳
‑
氧键,即c
‑
o键(包括醇衍生的c
‑
o键、醚衍生的c
‑
o键和环氧衍生的c
‑
o键)和c=o键(包括羰基衍生的c=o键、羧基衍生的c=o键、酯衍生的c=o键和内酯衍生的c=o键)的总量相对于所有键,即c
‑
c键、c=c键、c
‑
h键、c
‑
o键和c=o键的总量的比例(该比例由c1s xps分析确定)优选为10%以上,更优选20%以上,还更优选25%以上,并且该比例的上限优选为35%以下。当含碳物质的材料中c
‑
o键和c=o键的总量相对于c
‑
c键、c=c键、c
‑
h键、c
‑
o键和c=o键的总量的比例(该比例由c1s xps分析确定)落在上述范围时,含碳物质的材料不同于迄今已知的简单碳物质,可以是具有各种物性如溶解性的新型含碳物质的材料。
[0167]
(2)醚衍生的c
‑
o键(即,c
‑
o
‑
c键)和醇衍生的c
‑
o键(即,c
‑
oh键)的总量相对于所有碳
‑
氧键,即c
‑
o键(包括醇衍生的c
‑
o键、醚衍生的c
‑
o键和环氧衍生的c
‑
o键)和c=o键(包括羰基衍生的c=o键、羧基衍生的c=o键、酯衍生的c=o键和内酯衍生的c=o键)的总量的比例(该比例由c1s xps分析确定)优选为50%以上,更优选60%以上,还更优选65%以上,特别优选70%以上,最优选75%以上。该比例的上限优选为90%以下。当含碳物质的材料中醚衍生的c
‑
o键和醇衍生的c
‑
o键的总量相对于c
‑
o键和c=o键的总量的比例(该比例
由c1s xps分析确定)落在上述范围时,可以改进含碳物质的材料的碳物质部分的结构控制率,并且可以更精确地控制其结构。即,随着源自于碳物质的分解反应的c=o键的比例变大,分解反应受到抑制,因此这种含碳物质的材料可以说是具有更精确控制的结构的含碳物质的材料。
[0168]
(3)醚衍生的c
‑
o键(即,c
‑
o
‑
c键)和醇衍生的c
‑
o键(即,c
‑
oh键)的总量相对于所有键,即c
‑
c键、c=c键、c
‑
h键、c
‑
o键(包括醇衍生的c
‑
o键、醚衍生的c
‑
o键和环氧衍生的c
‑
o键)和c=o键(包括羰基衍生的c=o键、羧基衍生的c=o键、酯衍生的c=o键和内酯衍生的c=o键)的总量的比例(该比例由c1s xps分析确定)优选为15%以上,更优选17%以上,还更优选20%以上。该比例的上限优选为30%以下。当含碳物质的材料中醚衍生的c
‑
o键(即,c
‑
o
‑
c键)和醇衍生的c
‑
o键(即,c
‑
oh键)的总量相对于所有键的总量的比例(该比例由c1s xps分析确定)落在上述范围时,含碳物质的材料可以说是具有更精确控制的结构的含碳物质的材料。
[0169]
(4)方面:其中所有碳
‑
氧键,即c
‑
o键(包括醇衍生的c
‑
o键、醚衍生的c
‑
o键和环氧衍生的c
‑
o键)和c=o键(包括羰基衍生的c=o键、羧基衍生的c=o键、酯衍生的c=o键和内酯衍生的c=o键)的总量相对于所有键,即c
‑
c键、c=c键、c
‑
h键、c
‑
o键和c=o键的总量的比例(该比例由c1s xps分析确定)落在方面(1)中所述的范围;以及其中醚衍生的c
‑
o键(即,c
‑
o
‑
c键)和醇衍生的c
‑
o键(即,c
‑
oh键)的总量相对于所有碳
‑
氧键,即c
‑
o键(包括醇衍生的c
‑
o键、醚衍生的c
‑
o键和环氧衍生的c
‑
o键)和c=o键(包括羰基衍生的c=o键、羧基衍生的c=o键、酯衍生的c=o键和内酯衍生的c=o键)的总量的比例(该比例由c1s xps分析确定)落在方面(2)中所述的范围。根据这样的方面,可以进一步改进含碳物质的材料的碳物质部分的溶解性,并且可以进一步改进碳物质部分的结构控制率。另外,根据这样的方面,可以甚至更精确地控制含碳物质的材料的结构。
[0170]
(5)含碳物质的材料在其ir分析中优选不显示由c=o拉伸振动导致的在1,660cm
‑1~1,800cm
‑1范围内的峰。当在含碳物质的材料的ir分析中在1,660cm
‑1~1,800cm
‑1范围内未观察到由c=o拉伸振动导致的峰时,可以更精确地控制含碳物质的材料的结构。当在含碳物质的材料的ir分析中在1,660cm
‑1~1,800cm
‑1范围内未观察到由c=o拉伸振动导致的峰时可以更精确地控制含碳物质的材料的结构的原因如上所述。
[0171]
(6)含碳物质的材料中的碳物质是溶剂可溶的。
[0172]
通过将由
‑
h基和
‑
oh基形成h2o并脱离而发生的缩合反应(上述缩合反应(a))作为缩合反应的典型实例来进行说明。
[0173]
第二实施方案中的化合物(a)的一个实施方案(有时称为“实施方案(x)”)为具有由一个碳六元环结构形成的骨架的化合物(a1),或具有其中两个以上的碳六元环结构彼此结合和/或稠合的骨架的化合物(a2),并且
‑
oh基占对骨架的结构形成没有贡献的取代基数量的一半,而
‑
h基占另一半。
[0174]
在实施方案(x)中,可以采用以下两种情况中的任一种:
[0175]
(i)化合物(a)是具有由一个碳六元环结构形成的骨架的化合物(a1)的情况;和
[0176]
(ii)化合物(a)是具有两个以上的碳六元环结构彼此结合和/或稠合的骨架的化合物(a2)的情况。
[0177]
在实施方案(x)中,术语“对骨架结构形成没有贡献的取代基”是指对在情况(i)下
“
由一个碳六元环结构形成的骨架”或在情况(ii)下“两个以上的碳六元环结构彼此结合和/或稠合的骨架”的结构形成没有贡献的取代基。例如,作为情况(i),当具有由一个碳六元环结构形成的骨架的化合物(a1)由后述的化学式(a1
‑
1)表示时,对由一个碳六元环结构形成的骨架的结构形成没有贡献的取代基为6个
‑
oh基和6个
‑
h基,并且当具有由一个碳六元环结构形成的骨架的化合物(a1)由后述的化学式(a1
‑
2)表示时,对由一个碳六元环结构形成的骨架的结构形成没有贡献的取代基为3个
‑
oh基和3个
‑
h基。另外,例如,作为情况(ii),当具有两个以上的碳六元环结构彼此结合和/或稠合的骨架的化合物(a2)由后述的化学式(a2
‑
1)表示时,对两个以上的碳六元环结构彼此结合和/或稠合的骨架的结构形成没有贡献的取代基为6个
‑
oh基和6个
‑
h基。
[0178]
在实施方案(x)中,
‑
oh基占对具有由一个碳六元环结构形成的骨架的化合物(a1)的骨架结构形成没有贡献的取代基的数量的一半,而
‑
h基占另一半。另外,
‑
oh基占对具有两个以上的碳六元环结构彼此结合和/或稠和的骨架的化合物(a2)的骨架结构形成没有贡献的取代基的数量的一半,而
‑
h基占另一半。当化合物(a)具有这种取代基构型时,该化合物可以通过加热而有效地引起相同分子之间和/或不同分子之间的脱水反应。
[0179]
可以在本发明的效果不受损的程度采用任何适当的化合物作为在实施方案(x)中可以采用的化合物(a),只要该化合物为具有由一个碳六元环结构形成的骨架的化合物(a1),或具有两个以上的碳六元环结构彼此结合和/或稠合的骨架的化合物(a2),并且
‑
oh基占对骨架的结构形成没有贡献的取代基的数量的一半,而
‑
h基占另一半。这种化合物(a)的实例包括以下化合物。
[0180][0181]
在实施方案(x)中可以采用的化合物(a)中,优选间苯三酚(化合物(a1
‑
2))或六羟基三亚苯(hhtp)(化合物(a2
‑
1)),因为通过由
‑
h基和
‑
oh基形成h2o并且脱离而引起的缩合反应认为是容易发生的,并且因为该反应认为是易于在低温下进行的。
[0182]
第二实施方案中的化合物(a)的另一个实施方案(有时称为“实施方案(y)”)是选自具有由一个碳六元环结构形成的骨架的化合物(a1)和/或具有两个以上的碳六元环结构彼此结合和/或稠合的骨架的化合物(a2)中的两种以上,并且
‑
oh基占对化合物(a1)的骨架结构形成没有贡献的取代基的数量和对化合物(a2)的骨架结构形成没有贡献的取代基的数量之和的一半,而
‑
h基占另一半。
[0183]
在实施方案(y)中,可以采用以下三种情况中的任一种:
[0184]
(i)化合物(a)由选自各自具有由一个碳六元环结构形成的骨架的化合物(a1)中的两种以上形成的情况;
[0185]
(ii)化合物(a)由选自各自具有两个以上的碳六元环结构彼此结合和/或稠合的骨架的化合物(a2)中的两种以上形成的情况;和
[0186]
(iii)化合物(a)由选自各自具有由一个碳六元环结构形成的骨架的化合物(a1)中的一种以上、和选自各自具有两个以上的碳六元环结构彼此结合和/或稠合的骨架的化合物(a2)中的一种以上形成的情况。
[0187]
在实施方案(y)中,术语“对化合物(a1)的骨架的结构形成没有贡献的取代基的数量和对化合物(a2)的骨架的结构形成没有贡献的取代基的数量之和”具有以下含义。即,在情况(i)下,该术语是指通过在两种以上的化合物(a1)的每一个中对“由一个碳六元环结构形成的骨架”的结构形成没有贡献的取代基的数量累加而得到的数量。在情况(ii)下,该术语是指通过在两种以上的化合物(a2)的每一个中对“两个以上的碳六元环结构彼此结合和/或稠合的骨架”的结构形成没有贡献的取代基的数量累加而得到的数量。在情况(iii)下,该术语是指通过在一种以上的化合物(a1)的每一个中对“由一个碳六元环结构形成的骨架”的结构形成没有贡献的取代基的数量、和在一种以上的化合物(a2)的每一个中对“两个以上的碳六元环结构彼此结合和/或稠合的骨架”的结构形成没有贡献的取代基的数量累加而得到的数量。
[0188]
在实施方案(y)中,例如,作为情况(i),当两种以上的化合物(a1)由以下化学式(a1
‑
5)和以下化学式(a1
‑
6)表示时,在由化学式(a1
‑
5)表示的化合物中对由一个碳六元环结构形成的骨架的结构形成没有贡献的取代基为2个
‑
oh基和4个
‑
h基,并且在由化学式(a1
‑
6)表示的化合物中对由一个碳六元环结构形成的骨架的结构形成没有贡献的基团为4个
‑
oh基和2个
‑
h基。因此,取代基的总数如下:6个
‑
oh基和6个
‑
h基。另外,例如,作为情况(iii),当一种以上的化合物(a1)由以下化学式(a1
‑
5)和以下化学式(a1
‑
7)表示,并且一种以上的化合物(a2)各自由以下化学式(a2
‑
3)表示时,在由化学式(a1
‑
5)表示的化合物中对由一个碳六元环结构形成的骨架的结构形成没有贡献的取代基为2个
‑
oh基和4个
‑
h基,在由化学式(a1
‑
7)表示的化合物中对由一个碳六元环结构形成的骨架的结构形成没有贡献的取代基为6个
‑
oh基,并且在由化学式(a2
‑
3)表示的化合物中对两个以上的碳六元环结构彼此结合和/或稠合的骨架的结构形成没有贡献的取代基为2个
‑
oh基和6个
‑
h基。
[0189][0190]
当使用这种化合物(a)时,反应由其自身的脱水反应引起,而不需要任何的反应催化剂。因此,可以抑制化学反应的副产物和反应催化剂存在于碳物质中而成为致命的杂质,并且由此可以得到质量更高的碳物质。另外,当使用这种化合物(a)时,可以在相对温和的温度环境下得到碳物质,而无需使用任何可燃性气体。另外,这样的化合物(a)可具有如此高的反应性以至于不需要催化作用。
[0191]
第二实施方案中的化合物(a)的优选实施方案例如是在分子中具有三个以上的酚羟基的化合物。
[0192]
可以在本发明的效果不受损的程度采用任何适当的在分子中具有三个以上的酚羟基的化合物作为在分子中具有三个以上的酚羟基的化合物。
[0193]
在分子中具有三个以上的酚羟基的化合物中,与酚羟基键合的芳香环优选为烃芳香环。即使当与酚羟基键合的芳香环为杂芳环时,也可以发挥本发明的效果。然而,具有更稳定的环结构的烃芳香环可以使要得到的碳物质更稳定。杂芳环不像其环结构仅包含碳的烃芳香环一样,是指其环结构包含碳和碳以外的其他元素的芳香环。
[0194]
在分子中具有三个以上的酚羟基的化合物可以具有除酚羟基以外的取代基。可以在本发明的效果不受损的程度采用任何适当的取代基作为此类取代基。这种取代基优选单独为羟基,因为进一步改进了本发明的效果。即使当存在除羟基以外的取代基时,也可以发挥本发明的效果。然而,当不存在除羟基以外的取代基时,易于防止副反应,因此该化合物更容易转变成碳物质。用作除酚羟基以外的取代基的本文中使用的“羟基”是指非酚的羟基。当然,术语“取代基”是指取代氢基(
‑
h)的基团。
[0195]
可以在本发明的效果不受损的程度采用任何适当的元素作为用于形成在分子中具有三个以上的酚羟基的化合物的元素。这样的元素优选单独地为碳、氧和氢,因为改进了本发明的效果。即使当存在除碳、氧和氢以外的元素时,也可以发挥本发明的效果。然而,当不存在除碳、氧和氢以外的元素时,易于防止副反应,因此该化合物更容易转变成碳物质。
[0196]
在分子中具有三个以上的酚羟基的化合物的缩合反应温度优选落在200℃~450℃的范围,更优选落在200℃~400℃的范围,以便可以进一步发挥本发明的效果。因而,该
化合物可以有效地转变成碳物质。
[0197]
各自在分子中具有三个以上的酚羟基的化合物可以单独或以其组合使用。即使当使用两种以上的化合物时,其分子间发生缩合反应的温度也优选落在上述范围内。
[0198]
在分子中具有三个以上的酚羟基的化合物的实例包括由通式(1)~(11)表示的化合物。
[0199]
[0200]
在通式(1)~(11)的每一个中,x表示氢原子或羟基,并且三个以上的x均表示羟基(酚羟基)。
[0201]
本文中,术语“酚羟基”是指键合至芳香环的羟基。即,在通式(1)中,键合至芳香环的6个x中的三个以上均表示酚羟基。在通式(2)中,键合至芳香环的6个x中的三个以上均表示酚羟基。在通式(3)中,键合至芳香环的10个x中的三个以上均表示酚羟基。在通式(4)中,键合至芳香环的11个x中的3个以上均表示酚羟基。在通式(5)中,键合至芳香环的9个x中的三个以上均表示酚羟基。在通式(6)中,键合至芳香环的9个x中的三个以上均表示酚羟基。在通式(7)中,键合至芳香环的10个x中的三个以上均表示酚羟基。在通式(8)中,键合至芳香环的11个x中的三个以上均表示酚羟基。在通式(9)中,键合至芳香环的9个x中的三个以上均表示酚羟基。在通式(10)中,键合至芳香环的9个x中的三个以上均表示酚羟基。在通式(11)中,键合至芳香环的12个x中的三个以上均表示酚羟基。
[0202]
各自在分子中具有三个以上的酚羟基的化合物中,优选间苯三酚或六羟基三亚苯,因为由
‑
h基和
‑
oh基形成h2o并脱离而引起的缩合反应认为是容易发生的,并且因为反应认为是容易进行的,更优选间苯三酚。
[0203]
[化合物(a)的典型实施方案(第三实施方案)]
[0204]
化合物(a)的典型实施方案(第三实施方案)是同时采用第一实施方案和第二实施方案两者的实施方案。即,第三实施方案是通过加热分解而在芳香环上产生自由基的芳族化合物,并且是一个中性分子由两种以上的基团通过它们的缩合反应形成并脱离这样的化合物。这样的化合物(a)可能导致相同分子之间和/或不同分子之间的缩合反应从而转变成碳物质。
[0205]
第三实施方案的具体结构例如为化合物(a3
‑
1)。二氧化碳分子通过加热将从化合物(a3
‑
1)中脱离出,在其芳香环上产生自由基(反应活性位点)。另外,羟基和氢基在化合物的分子之间经历脱水而引起缩合反应。
[0206][0207]
当使用这种芳族化合物时,反应仅由其自身的脱水反应引起,而不需要任何反应催化剂。因此,可以抑制化学反应的副产物和反应催化剂存在于碳物质中而成为致命的杂质,并由此可以得到品质更高的碳物质。另外,当使用这种化合物(a)时,可以在相对温和的温度环境下得到碳物质,而无需使用任何可燃性气体。另外,这样的化合物(a)可具有如此
高的反应性以至于不需要催化作用。
[0208]
<<1
‑
2.无机物>>
[0209]
可以在本发明的效果不受损的程度采用任何适当的无机物作为无机物。例如,可以采用颗粒状无机物(无机物颗粒)或非颗粒状无机物(例如纤维状无机物或薄膜状无机物)作为这种无机物。无机物优选为颗粒状无机物(无机物颗粒)。
[0210]
无机物可以单独或以其组合使用。
[0211]
无机物优选为例如选自由无机氧化物、无机氮化物、无机硫化物、无机碳化物和不溶性盐组成的组中的至少一种。
[0212]
部分氧化的金属,或者优选地,其表面至少部分氧化的金属也包括在本发明中使用的“无机氧化物”的实例中。这是因为将金属的一部分,或者优选地,其表面的至少一部分通常如后所述氧化。此类金属优选为易于氧化的金属,例如镁(mg)、钙(ca)、锶(sr)、钡(ba)、钛(ti)、锆(zr)、铪(hf)、钒(v)、铌(nb)、钽(ta)、铬(cr)、钼(mo)、钨(w)、锰(mn)、铟(in)、镓(ga)、铁(fe)、钴(co)、镍(ni)、铜(cu)、锌(zn)、镉(cd)、铝(al)、锡(sn)、镧(la)、钇(y)、铈(ce)或硅(si),更优选铜(cu)、铝(al)或硅(si)。即,任何这样的金属都容易被氧化,因此将金属的一部分,或者优选地,其表面的至少一部分氧化。因此,金属包括在本发明中使用的“无机氧化物”的类别中。
[0213]
本发明中使用的“无机氧化物”的具体实例包括二氧化硅、氧化铝、二氧化钛、多酸、被部分氧化的金属(优选其表面被至少部分氧化的金属)/复合氧化物和固溶体氧化物。即,本发明中使用的“无机氧化物”可以是包含一种金属元素的氧化物,可以是包含两种以上的金属元素的复合氧化物,或者可以是所谓的固溶体氧化物,其中异种元素进一步固溶在包括一种金属元素(也称为“单一金属氧化物”)或复合氧化物的氧化物中。固溶体氧化物中的异种元素可以是金属元素,或者可以是除氧以外的非金属元素,例如氮或氟。
[0214]
无机氧化物可以单独或以其组合使用。采用两种以上的无机氧化物的实例例如是仅将两种以上的无机氧化物组合使用(例如混合)的情况,或者将两种以上的无机氧化物相互结合的情况。
[0215]“包含一种金属元素的氧化物”的实例包括氧化镁、氧化钙、氧化锶、氧化钡、氧化钛、氧化锆、氧化铪、氧化钒、氧化铌、氧化钽、氧化铬、氧化钼、氧化钨、氧化锰、氧化铟、氧化镓、氧化铁、氧化钴、氧化镍、氧化铜、氧化锌、氧化镉、氧化铝、氧化锡、氧化镧、氧化钇、氧化铈和氧化硅。其中,优选氧化镁、氧化钛(二氧化钛)、氧化铝(三氧化二铝)和氧化硅(二氧化硅)。
[0216]
可以在本发明的效果不受损的程度采用任何适当的复合氧化物作为上述“包含两种以上的金属元素的复合氧化物”。这种复合氧化物典型地是包含两种以上的金属的氧化物,并且其实例包括钙钛矿结构的复合氧化物和尖晶石结构的复合氧化物。
[0217]
钙钛矿结构的复合氧化物典型地是由abo3表示的氧化物(a和b表示彼此不同的元素),其实例包括钙钛矿(catio3)、钛酸钡(batio3)、钛酸锶(srtio3)、钛酸锆酸铅(pb(zr,ti)o3)、锆酸钡(bazro3)和铌酸锂(linbo3)。
[0218]
尖晶石结构的复合氧化物的实例包括尖晶石(mgal2o4)、钛酸锂(liti2o4)和金绿玉石(beal2o4)。
[0219]
可以在本发明的效果不受损的程度采用任何适当的固溶体氧化物作为上述“固溶
体氧化物”。这种固溶体氧化物典型地是通过将异种金属元素和/或除氧以外的非金属元素例如氮或氟固溶在单一金属氧化物或复合氧化物中而得到的产物。
[0220]
本发明中使用的“无机氧化物”的形状不受限制(即,例如,氧化物可以是无机氧化物颗粒或可以是非颗粒状无机氧化物)。允许完全为无机氧化物的实施方案,并且也允许部分为无机氧化物的实施方案。部分为无机氧化物的实施方案优选为在其表面具有无机氧化物的实施方案。
[0221]
对部分为无机氧化物的实施方案(优选在其表面上具有无机氧化物的实施方案)的形状没有限制,其实例为被部分氧化的金属(优选为其表面被至少部分氧化的金属)。金属的一部分(优选其表面的至少一部分)可以在氧气存在下被氧化。因此,作为本发明中使用的无机氧化物的一个实施方案的“在其表面具有无机氧化物的实施方案”的形状没有限定,并且其表面被至少部分氧化的金属也包括在其类别中。
[0222]
在使用无机氧化物颗粒作为无机氧化物的情况下,典型地可以通过加热含有化合物(a)和无机氧化物的组合物的加热步骤(i)来获得包含许多无机氧化物颗粒的块状含碳物质的材料(在这种情况下,可以通过切碎等得到颗粒状含碳物质的材料)。然而,颗粒状含碳物质的材料可以借助于调节化合物(a)与无机氧化物之间的共混比,通过加热含有化合物(a)和无机氧化物的组合物的加热步骤(i)来得到。
[0223]
当使用纤维状无机氧化物作为无机氧化物时,例如,可以代替后述的核壳颗粒而得到纤维状的核壳纤维。另外,当使用纤维状无机氧化物时,例如,可以代替后述的中空碳细颗粒而得到管状的中空碳物质。
[0224]
当将薄膜状无机氧化物用作无机氧化物时,例如,层压的含碳物质的材料可以通过加热含有化合物(a)和无机氧化物的组合物的加热步骤(i)来得到。另外,例如,各种薄膜状碳物质可以通过进一步使这样的层压的含碳物质的材料经受例如后述的加热步骤(ii)、碳物质除去步骤或无机氧化物除去步骤来得到。
[0225]
无机氧化物的分解温度优选为800℃以上,更优选850℃以上,还更优选900℃以上,特别优选950℃以上,因为可以进一步表现出本发明的效果。
[0226]
本发明中,无机氧化物优选为无机氧化物颗粒,因为可以进一步表现出本发明的效果。
[0227]
本文中使用的“无机氧化物颗粒”优选是完全为无机氧化物的颗粒或部分为无机氧化物的颗粒。部分为无机氧化物的颗粒优选是各自在其表面上具有无机氧化物的颗粒。本文中使用的“无机氧化物颗粒”更优选是完全为无机氧化物的颗粒。
[0228]
部分为无机氧化物的颗粒(优选各自在其表面上具有无机氧化物的颗粒)例如是被部分氧化的金属颗粒(优选其表面被至少部分氧化的金属颗粒)。金属颗粒的一部分(优选其表面的至少一部分)可以在氧气的存在下被氧化。因此,其表面被至少部分地氧化的金属颗粒包括在作为本发明中使用的无机氧化物颗粒的一个实施方案的“各自在其表面上具有无机氧化物的颗粒”的类别中。
[0229]
除了金属颗粒以外的无机氧化物颗粒(例如,二氧化硅颗粒、氧化铝颗粒和二氧化钛颗粒)不仅可以是完全为无机氧化物的颗粒,而且可以是部分为无机氧化物的颗粒(优选各自在其表面上具有无机氧化物的颗粒)。即,例如,当以二氧化硅颗粒为例时,二氧化硅颗粒不仅可以是完全为二氧化硅的颗粒,而且可以是部分为二氧化硅的颗粒(优选各自在其
表面上具有二氧化硅的颗粒)。
[0230]
无机氧化物颗粒的平均粒径可以根据目的适当设定。无机氧化物颗粒的平均粒径优选为0.01μm~100μm,特别优选0.1μm~10μm,因为可以进一步表现出本发明的效果。
[0231]
无机氧化物颗粒的平均粒径是在基于体积的粒径分布中的平均粒径,并且优选通过激光衍射散射法测量。
[0232]
无机氧化物颗粒优选为各自在其表面上具有官能团的无机氧化物颗粒,因为可以进一步表现出本发明的效果。此类官能团的实例包括:羟基官能团,例如m
‑
oh;含有醚官能团的氧官能团,例如m
‑
o
‑
m;含有氨基官能团的氮官能团,例如m
‑
nh2或m
‑
nh
‑
m;含有巯基官能团的硫官能团,例如m
‑
sh或m
‑
s
‑
m;以及包括硅官能团、硼官能团和磷官能团的其他官能团(其中m概念性地表示与官能团键合的对象,并且表示可以与官能团键合的任何适当的对象,例如无机氧化物本身,例如,用于形成无机氧化物的金属元素或有机基团)。这些官能团可以容易地通过例如用各种化合物处理无机氧化物的表面来形成。其中,作为无机氧化物颗粒,优选各自在其表面上具有氧官能团的无机氧化物颗粒。
[0233]
当采用各自在其表面上具有官能团的无机氧化物颗粒作为无机氧化物时,存在于各无机氧化物颗粒的表面上的官能团可以与含碳物质的材料中的碳物质形成键。有助于这种键的碳物质的区域(碳物质结合区域)不是碳物质本身。即,如图1所示,在含碳物质的材料的一个优选实施方案中,有机无机复合体(其细节将在后面描述)100通过以下来得到:将多个无机物颗粒20(在这种情况下,无机氧化物颗粒)分散在作为基体的碳物质10中,并且在碳物质10与各无机物颗粒20之间的界面处,存在通过在碳物质和各无机物颗粒20的表面上存在的官能团之间形成键而产生的碳物质区域(碳物质结合区域)30。然后,例如,当碳物质可溶于溶剂中时,用溶解碳物质的溶剂处理有机无机复合体100可以提供核壳颗粒(核部分:无机氧化物颗粒,壳部分:碳物质结合区域)200,其中如图2所示,无机物颗粒20的表面被覆有碳物质结合区域(未被溶剂溶解的区域)30。从如此得到的核壳颗粒200中除去用作核部分的无机物颗粒20可以提供具有碳物质的中空碳细颗粒。
[0234]
各自在其表面上具有官能团的无机氧化物颗粒优选为选自由二氧化硅颗粒、氧化铝颗粒、二氧化钛颗粒、氧化镁颗粒、多酸颗粒、其表面被至少部分地氧化的金属颗粒、复合氧化物颗粒、和固溶体氧化物颗粒组成的组中的至少一种。当采用选自由二氧化硅颗粒、氧化铝颗粒、二氧化钛颗粒、氧化镁颗粒、多酸颗粒、其表面被至少部分地氧化的金属颗粒、复合氧化物颗粒、和固溶体氧化物颗粒组成的组中的至少一种作为各自在其表面上具有官能团的无机氧化物颗粒时,在各无机氧化物颗粒的表面上存在的官能团可以进一步与碳物质形成键。
[0235]
各自在其表面上具有官能团的无机氧化物颗粒更优选为多酸颗粒。用于形成多酸颗粒的多酸例如为同多酸或杂多酸。如上所述,当使用化合物(a)时,可以得到含碳物质的材料,而无需利用任何催化效果。然而,特别地,当采用多酸颗粒作为各自在其表面上具有官能团的无机氧化物颗粒时,强酸性催化效果和表面官能团的存在彼此配合。因而,在该化合物转变成含碳物质的材料的情况下,其分子之间的脱水缩合反应加速,增加
‑
oh基和
‑
o
‑
基的总量相对于c
‑
o键的总量的比例。因而,含碳物质的材料可以具有高的结构控制率。
[0236]
作为同多酸,可以在本发明的效果不受损的程度采用任何适当的同多酸。这种同多酸的实例包括主要由无机元素形成的无机酸,所述无机元素例如钼、钒、钨、铌、钛、钽、
铬、锰、铼、铁、钌、钴、镍、钯、铂、铜、银、金、锡、钛、锆、铑、铱、锇或锌,及其盐。其典型实例包括钼酸、钒酸、钨酸、铌酸、钛酸和钽酸。
[0237]
作为杂多酸,可以在本发明的效果不受损的程度采用任何适当的杂多酸。这种杂多酸的实例是通过将杂原子引入同多酸或其金属盐而形成的杂多酸。杂原子的实例包括氧、硫、磷、铵、钾、钠和硅。杂多酸可以是水合物。
[0238]
杂多酸的具体实例包括通过将杂原子引入到含钨的同多酸中形成的钨系杂多酸,和通过将杂原子引入到含钼的同多酸中形成的钼系杂多酸。
[0239]
钨系杂多酸的实例包括磷钨酸、硅钨酸、钴钨酸、锗钨酸、硼钨酸、磷钒钨酸和磷钨钼酸。
[0240]
钼系杂多酸的实例包括磷钼酸、硅钼酸和磷钒钼酸。
[0241]
作为无机氮化物,可以在本发明的效果不受损的程度采用任何适当的无机氮化物。这种无机氮化物的实例包括氮化硼、氮化碳、氮化铝和氮化镓。其中,优选氮化硼和氮化铝。
[0242]
无机氮化物可以单独或以其组合使用。
[0243]
本发明中,如前述的无机氧化物中一样,无机氮化物颗粒优选作为无机氮化物,因为可以进一步表现出本发明的效果。
[0244]
无机氮化物颗粒的平均粒径可以根据目的适当设定。无机氮化物颗粒的平均粒径优选为0.01μm~100μm,特别优选0.1μm~10μm,因为可以进一步表现出本发明的效果。
[0245]
无机氮化物颗粒的平均粒径是在基于体积的粒径分布中的平均粒径,并且优选通过激光衍射散射法测量。
[0246]
无机氮化物颗粒优选为各自在其表面上具有官能团的无机氮化物颗粒,因为可以进一步表现出本发明的效果。此类官能团的实例包括:羟基官能团,例如m
‑
oh;含有醚官能团的氧官能团,例如m
‑
o
‑
m;含有氨基官能团的氮官能团,例如m
‑
nh2或m
‑
nh
‑
m;含有巯基官能团的硫官能团,例如m
‑
sh或m
‑
s
‑
m;以及包括硅官能团、硼官能团和磷官能团的其他官能团(其中m概念性地表示与官能团键合的对象,并且表示可以与官能团键合的任何适当的对象,例如无机氮化物本身,例如,用于形成无机氮化物的金属元素或有机基团)。这些官能团可以容易地通过例如用各种化合物处理无机氮化物的表面来形成。
[0247]
当采用各自在其表面上具有官能团的无机氮化物颗粒作为无机氮化物时,存在于各无机氮化物颗粒的表面上的官能团可以与含碳物质的材料中的碳物质形成键。有助于这种键的碳物质的区域(碳物质结合区域)不是碳物质本身。即,像前述中的各自在其表面上具有官能团的无机氧化物颗粒一样,如图1所示,在含碳物质的材料的一个优选实施方案中,有机无机复合体(其细节将在后面描述)100通过以下来得到:将多个无机物颗粒20(在这种情况下,无机氮化物颗粒)分散在作为基体的碳物质10中,并且在碳物质10与各无机物颗粒20之间的界面处,存在通过在碳物质和各无机物颗粒20的表面上存在的官能团之间形成键而产生的碳物质区域(碳物质结合区域)30。然后,例如,当碳物质可溶于溶剂中时,用溶解碳物质的溶剂处理有机无机复合体100可以提供核壳颗粒(核部分:无机氮化物颗粒,壳部分:碳物质结合区域)200,其中如图2所示,无机物颗粒20的表面被覆有碳物质结合区域(未被溶剂溶解的区域)30。从如此得到的核壳颗粒200中除去用作核部分的无机物颗粒20可以提供具有碳物质的中空碳细颗粒。
[0248]
作为无机硫化物,可以在本发明的效果不受损的程度采用任何适当的无机硫化物。这种无机硫化物的实例包括硫化铜、硫化锌和硫化镉。
[0249]
无机硫化物可以单独或以其组合使用。
[0250]
本发明中,如前述的无机氧化物中一样,无机硫化物颗粒优选为无机硫化物,因为可以进一步表现出本发明的效果。
[0251]
无机硫化物颗粒的平均粒径可以根据目的适当设定。无机硫化物颗粒的平均粒径优选为0.01μm~100μm,特别优选0.1μm~10μm,因为可以进一步表现出本发明的效果。
[0252]
无机硫化物颗粒的平均粒径是在基于体积的粒径分布中的平均粒径,并且优选通过激光衍射散射法测量。
[0253]
无机硫化物颗粒优选为各自在其表面上具有官能团的无机硫化物颗粒,因为可以进一步表现出本发明的效果。此类官能团的实例包括:羟基官能团,例如m
‑
oh;含有醚官能团的氧官能团,例如m
‑
o
‑
m;含有氨基官能团的氮官能团,例如m
‑
nh2或m
‑
nh
‑
m;含有巯基官能团的硫官能团,例如m
‑
sh或m
‑
s
‑
m;以及包括硅官能团、硼官能团和磷官能团的其他官能团(其中m概念性地表示与官能团键合的对象,并且表示可以与官能团键合的任何适当的对象,例如无机硫化物本身,例如,用于形成无机硫化物的金属元素或有机基团)。这些官能团可以容易地通过例如用各种化合物处理无机硫化物的表面来形成。
[0254]
当采用各自在其表面上具有官能团的无机硫化物颗粒作为无机硫化物时,存在于各无机硫化物颗粒的表面上的官能团可以与含碳物质的材料中的碳物质形成键。有助于这种键的碳物质的区域(碳物质结合区域)不是碳物质本身。即,像前述中的各自在其表面上具有官能团的无机氧化物颗粒一样,如图1所示,在含碳物质的材料的一个优选实施方案中,有机无机复合体(其细节将在后面描述)100通过以下来得到:将多个无机物颗粒20(在这种情况下,无机硫化物颗粒)分散在作为基体的碳物质10中,并且在碳物质10与各无机物颗粒20之间的界面处,存在通过在碳物质和各无机物颗粒20的表面上的官能团之间形成键而产生的碳物质区域(碳物质结合区域)30。然后,例如,当碳物质可溶于溶剂中时,用溶解碳物质的溶剂处理有机无机复合体100可以提供核壳颗粒(核部分:无机硫化物颗粒,壳部分:碳物质结合区域)200,其中如图2所示,无机物颗粒20的表面被覆有碳物质结合区域(未被溶剂溶解的区域)30。从如此得到的核壳颗粒200中除去用作核部分的无机物颗粒20可以提供具有碳物质的中空碳细颗粒。
[0255]
作为无机碳化物,可以在本发明的效果不受损的程度采用任何适当的无机碳化物。这种无机碳化物的实例包括碳化硅、碳化钨和碳化钙。
[0256]
无机碳化物可以单独或以其组合使用。
[0257]
本发明中,如前述的无机氧化物中一样,无机碳化物颗粒优选为无机碳化物,因为可以进一步表现出本发明的效果。
[0258]
无机碳化物颗粒的平均粒径可以根据目的适当设定。无机碳化物颗粒的平均粒径优选为0.01μm~100μm,特别优选0.1μm~10μm,因为可以进一步表现出本发明的效果。
[0259]
无机碳化物颗粒的平均粒径是在基于体积的粒径分布中的平均粒径,并且优选通过激光衍射散射法测量。
[0260]
无机碳化物颗粒优选为各自在其表面上具有官能团的无机碳化物颗粒,因为可以进一步表现出本发明的效果。此类官能团的实例包括:羟基官能团,例如m
‑
oh;含有醚官能
团的氧官能团,例如m
‑
o
‑
m;含有氨基官能团的氮官能团,例如m
‑
nh2或m
‑
nh
‑
m;含有巯基官能团的硫官能团,例如m
‑
sh或m
‑
s
‑
m;以及包括硅官能团、硼官能团和磷官能团的其他官能团(其中m概念性地表示与官能团键合的对象,并且表示可以与官能团键合的任何适当的对象,例如无机碳化物本身,例如,用于形成无机碳化物的金属元素或有机基团)。这些官能团可以容易地通过例如用各种化合物处理无机碳化物的表面来形成。
[0261]
当采用各自在其表面上具有官能团的无机碳化物颗粒作为无机碳化物时,存在于各无机碳化物颗粒的表面上的官能团可以与含碳物质的材料中的碳物质形成键。有助于这种键的碳物质的区域(碳物质结合区域)不是碳物质本身。即,像前述中的各自在其表面上具有官能团的无机氧化物颗粒一样,如图1所示,在含碳物质的材料的一个优选实施方案中,有机无机复合体(其细节将在后面描述)100通过以下来得到:将多个无机物颗粒20(在这种情况下,无机碳化物颗粒)分散在作为基体的碳物质10中,并且在碳物质10与各无机物颗粒20之间的界面处,存在通过在碳物质和各无机物颗粒20的表面上存在的官能团之间形成键而产生的碳物质区域(碳物质结合区域)30。然后,例如,当碳物质可溶于溶剂中时,用溶解碳物质的溶剂处理有机无机复合体100可以提供核壳颗粒(核部分:无机碳化物颗粒,壳部分:碳物质结合区域)200,其中如图2所示,无机物颗粒20的表面被覆有碳物质结合区域(未被溶剂溶解的区域)30。从如此得到的核壳颗粒200中除去用作核部分的无机物颗粒20可以提供具有碳物质的中空碳细颗粒。
[0262]
作为不溶性盐,可以在本发明的效果不受损的程度采用任何适当的不溶性盐。这种不溶性盐优选为在有机溶剂中不可溶的含金属盐。其实例包括金属磷酸盐,例如磷酸铁锂,和金属硫酸盐。其中,优选为磷酸铁锂。
[0263]
不溶性盐可以单独或以其组合使用。
[0264]
不溶性盐仅需要在本发明的实施方案中的含碳物质的材料的制造方法中要使用的溶剂中是不可溶的。例如,当在混合化合物(a)和无机物的步骤中使用溶剂时,无机物仅需要在溶剂中是不可溶的。换言之,不溶解无机物的适当的溶剂仅需要根据无机物来选择。在本发明的实施方案中的含碳物质的材料的制造方法包括碳物质除去步骤的情况下对于碳物质除去步骤同样如此。在本发明的实施方案中在含碳物质的材料的制造方法中当制造中空碳细颗粒等时,该方法优选包括溶解无机物的步骤。然而,即使当该方法包括这种溶解无机物的步骤,也可以在该步骤之前的步骤中选择不溶解在溶剂中的无机物。术语“不溶性盐”是指该盐可以在一定条件下溶解于溶剂中,但是在可以发挥本发明的效果的制造方法中不溶于溶剂中。另外,不溶性盐也优选为颗粒状形式,其平均粒径的优选范围与氧化物的是相同的。
[0265]
当采用选自由二氧化硅颗粒和多酸颗粒组成的组中的至少一种作为各自在其表面上具有官能团的无机氧化物颗粒时,存在于各无机氧化物颗粒的表面上的官能团可以进一步地与碳物质形成键,因此要得到的含碳物质的材料优选特别具有以下方面(4)。
[0266]
(4)含碳物质的材料在其ir分析中优选不显示在1,660cm
‑1~1,800cm
‑1的范围内由c=o拉伸振动导致的峰。当在含碳物质的材料的ir分析中在1,660cm
‑1~1,800cm
‑1范围内未观察到由c=o拉伸振动导致的峰在1,660cm
‑
1~1,800cm
‑1的范围内时,可以更精确地控制含碳物质的材料的结构。
[0267]
<<<<2.含碳物质的材料的制造方法的典型实例>>>>
[0268]
可以通过本发明的含碳物质的材料的制造方法制造的含碳物质的材料种类繁多,并且可以采用诸如颗粒状和非颗粒状(例如,纤维状或薄膜状)等的各种形状中的任一种作为其形状。形状优选为颗粒状。
[0269]
如上所述,例如,当分别采用各种形状的无机物,例如颗粒状无机物(无机物颗粒)和非颗粒状无机物(例如纤维状无机物或薄膜状无机物)作为无机物时,多种多样的含碳物质的材料可以根据各自的形状来得到。无机物优选为颗粒状无机物(无机物颗粒)。
[0270]
在使用无机物颗粒的情况下,包含许多无机物颗粒的块状的含碳物质的材料可以典型地通过本发明的含碳物质的材料的制造方法来得到。在这种情况下,可以通过切碎等得到颗粒状含碳物质的材料。另外,可以借助于本发明的含碳物质的材料的制造方法,通过调节化合物(a)与无机物之间的共混比例来得到颗粒状含碳物质的材料。
[0271]
当使用纤维状无机氧化物时,通过本发明的含碳物质的材料的制造方法,可以代替后述的核壳颗粒而得到纤维状的核壳纤维。另外,当使用纤维状无机物时,通过本发明的含碳物质的材料的制造方法,可以代替后述的中空碳细颗粒而得到管状的中空碳物质。
[0272]
当使用薄膜状无机物时,通过本发明的含碳物质的材料的制造方法,可以得到层压的含碳物质的材料。另外,各种薄膜状碳物质可以通过进一步将这种层压的含碳物质的材料进行例如后述的加热步骤(ii)、碳物质除去步骤或无机物除去步骤来得到。
[0273]
当控制可以通过本发明的含碳物质的材料的制造方法制造的含碳物质的材料中的碳物质部分的厚度时,含碳物质的材料可以采取各种的用途。这种碳物质部分的厚度通过例如将其以下特定方面作为典型实例进行描述:
[0274]
(方面1)在含碳物质的材料中通过碳物质除去步骤将除了与无机物的最外表面强烈相互作用的碳物质之外的存在于无机物表面的碳物质全体除去而得到的碳物质部分的厚度;和
[0275]
(方面2)在含碳物质的材料的碳物质部分的全体或一部分剩余的状态下,在含碳物质的材料中的碳物质部分的厚度。
[0276]
上述(方面1)中的碳物质部分更具体地是与无机物的最外表面强烈相互作用的碳物质部分,该部分是通过碳物质除去步骤将除了与无机物的最外表面强烈相互作用的碳物质之外的存在于无机物表面的碳物质全体除去而得到的。这种碳物质部分的厚度小,并且优选为0.3nm~10nm,更优选0.4nm~3nm,尽管优选的值根据碳物质的结构而变化。当将碳物质部分的厚度控制在这样的范围内时,可以在各种用途中充分利用与无机物强烈相互作用的碳组分。
[0277]
在上述(方面1)中,碳物质部分的厚度典型地是小的,但是厚度可以通过使用适当的碳物质除去方法来调节而没有任何上限。
[0278]
尽管上述(方面2)中的碳物质部分的厚度通常大于上述(方面1)中的碳物质部分的厚度,但是该厚度优选为100μm以下,更优选50μm以下,还更优选10μm以下,最优选1μm以下,以便可以进一步发挥其作为碳物质膜的功能。当将上述(方面2)中的碳物质部分的厚度控制在这样的范围内时,可以充分发挥其作为膜的功能。
[0279]
碳物质部分的厚度也可以根据用于含碳物质的材料的制造方法中的原料化合物(例如,化合物(a))的种类和制造条件来适当地控制。
[0280]
这种碳物质部分的厚度可以通过各种分析方法来确定。这样的分析方法的实例包
括:包括用电子显微镜直接观察该部分的方法(方法a);包括通过元素分析或热重分析计算出的,从无机物的宏观表面积(不包括像本发明的有机分子不能进入的微结构的表面积)和碳物质的密度估算碳量的方法(方法b)。
[0281]
上述(方法a)更具体地例如是包括用透射电子显微镜或扫描电子显微镜,优选安装有能量色散x射线分析仪的透射电子显微镜或扫描电子显微镜观察作为样品的含碳物质的材料的截面的方法。扫描电子显微镜。
[0282]
在上述(方法b)中,无机物的宏观表面积可以从例如作为样品的含碳物质的材料的形状和要掺入其中的无机物的形状来估算。
[0283]
在上述(方法a)中,例如,当样品是颗粒时,可以如下得到样品的平均厚度:优选在3处以上的位置处测量样品中单个颗粒的厚度,并采用其简单的平均值作为颗粒的厚度;更优选地对于10个以上的颗粒确定厚度,并且采用其简单的平均值作为平均厚度。如此得到的厚度(平均厚度)优选例如落在上述期望的厚度范围内,在该范围内希望控制碳物质部分的厚度。
[0284]
在上述(方法b)中,分析的厚度可以认为是样品的平均厚度。通过该(方法b)分析出的厚度优选例如落在上述期望的厚度范围内,在该范围内希望控制碳物质部分的厚度。
[0285]
这种含碳物质的材料的典型实施方案例如是有机无机复合体和含碳物质的颗粒。含碳物质的颗粒的实例包括核壳颗粒、高度碳化的核壳颗粒、中空碳细颗粒和高度碳化的中空碳细颗粒。下面描述典型且特定的含碳物质的材料的制造方法。
[0286]
<<2
‑
1.有机无机复合体的制造方法>>
[0287]
通过本发明的制造方法得到的含碳物质的材料的一个实施方案是有机无机复合体。
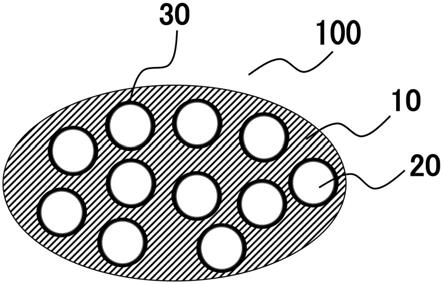
[0288]
有机无机复合体可以典型地通过加热含有化合物(a)和无机物的组合物的加热步骤(i)而得到,该化合物(a)通过加热而引起相同分子之间和/或不同分子之间的缩合反应。
[0289]
有机无机复合体包含碳物质和无机物。碳物质可以典型地通过加热化合物(a)来制造,在含有化合物(a)和无机物的组合物中通过加热该组合物的加热步骤(i),该化合物(a)通过加热而引起相同分子之间和/或不同分子之间的缩合反应。
[0290]
碳物质可以单独或以其组合使用。无机物可以单独或以其组合使用。<<1
‑
2.无机物>>章节中的说明可以援引至“无机物”中。
[0291]
有机无机复合体中碳物质的含量以重量比计优选为0.01wt%~99.99wt%,特别优选0.1wt%~99.9wt%。当有机无机复合体中的碳物质的含量落在上述范围时,可以在温和条件下工业地制造有机无机复合体,并且可以通过使用有机无机复合体作为材料来工业地制造中空碳细颗粒等。这种碳物质的含量可以根据目标物性通过例如稍后将描述的各种除去步骤而容易地设定为任何适当的比例。
[0292]
有机无机复合体中无机物的含量以重量比计优选为0.01wt%~99.99wt%,特别优选0.1wt%~99.9wt%。当有机无机复合体中的无机物的含量落在上述范围时,可以在温和条件下工业地制造有机无机复合体,并且可以通过使用有机无机复合体作为材料来工业地制造中空碳细颗粒等。这种无机物的含量可以根据目标物性通过例如稍后将描述的各种除去步骤而容易地设定为任何适当的比例。
[0293]
有机无机复合体中所有碳
‑
氧键,即c
‑
o键(包括醇衍生的c
‑
o键、醚衍生的c
‑
o键和
环氧衍生的c
‑
o键)和c=o键(包括羰基衍生的c=o键、羧基衍生的c=o键、酯衍生的c=o键和内酯衍生的c=o键)的总量相对于所有键,即c
‑
c键、c=c键、c
‑
h键、c
‑
o键和c=o键的总量的比例(该比例由c1s xps分析确定)优选为10%以上,更优选20%以上,还更优选25%以上。该比例的上限优选为35%以下。当有机无机复合体中c
‑
o键和c=o键的总量相对于c
‑
c键、c=c键、c
‑
h键、c
‑
o键和c=o键的总量的比例(该比例由c1s xps分析确定)落在上述范围时,有机无机复合体不同于迄今已知的简单碳物质,可以是具有各种物性如溶解性的新型含碳物质的材料。
[0294]
有机无机复合体中醚衍生的c
‑
o键(即,c
‑
o
‑
c键)和醇衍生的c
‑
o键(即,c
‑
oh键)的总量相对于所有碳
‑
氧键,即c
‑
o键(包括醇衍生的c
‑
o键、醚衍生的c
‑
o键和环氧衍生的c
‑
o键)和c=o键(包括羰基衍生的c=o键、羧基衍生的c=o键、酯衍生的c=o键和内酯衍生的c=o键)的总量的比例(该比例由c1s xps分析确定)优选为50%以上,更优选60%以上,还更优选65%以上,特别优选70%以上,最优选75%以上。该比例的上限优选为90%以下。当有机无机复合体中醚衍生的c
‑
o键和醇衍生的c
‑
o键的总量相对于c
‑
o键和c=o键的总量的比例(该比例由c1s xps分析确定)落在上述范围时,可以改进有机无机复合体的碳物质部分的结构控制率,并且可以更精确地控制其结构。即,随着源自于有机无机复合体的分解反应的c=o键的比例变小,分解反应受到抑制,因此这种有机无机复合体可以说是具有更精确控制的结构的含碳物质的材料。
[0295]
有机无机复合体中醚衍生的c
‑
o键(即,c
‑
o
‑
c键)和醇衍生的c
‑
o键(即,c
‑
oh键)的总量相对于所有键,即c
‑
c键、c=c键、c
‑
h键、c
‑
o键(包括醇衍生的c
‑
o键、醚衍生的c
‑
o键和环氧衍生的c
‑
o键)和c=o键(包括羰基衍生的c=o键、羧基衍生的c=o键、酯衍生的c=o键和内酯衍生的c=o键)的总量的比例(该比例由c1s xps分析确定)优选为15%以上,更优选17%以上,还更优选20%以上。该比例的上限优选为30%以下。当含碳物质的材料中醚衍生的c
‑
o键(即,c
‑
o
‑
c键)和醇衍生的c
‑
o键(即,c
‑
oh键)的总量相对于所有键的总量的比例(该比例由c1s xps分析确定)落在上述范围时,有机无机复合体可以说是具有更精确控制的结构的有机无机复合体。
[0296]
有机无机复合体特别优选为以下方面:其中所有碳
‑
氧键,即c
‑
o键(包括醇衍生的c
‑
o键、醚衍生的c
‑
o键和环氧衍生的c
‑
o键)和c=o键(包括羰基衍生的c=o键、羧基衍生的c=o键、酯衍生的c=o键和内酯衍生的c=o键)的总量相对于所有键,即c
‑
c键、c=c键、c
‑
h键、c
‑
o键和c=o键的总量的比例(该比例由c1s xps分析确定)落在上述范围;以及其中醚衍生的c
‑
o键(即,c
‑
o
‑
c键)和醇衍生的c
‑
o键(即,c
‑
oh键)的总量相对于所有碳
‑
氧键,即c
‑
o键(包括醇衍生的c
‑
o键、醚衍生的c
‑
o键和环氧衍生的c
‑
o键)和c=o键(包括羰基衍生的c=o键、羧基衍生的c=o键、酯衍生的c=o键和内酯衍生的c=o键)的总量的比例(该比例由c1s xps分析确定)落在上述范围。根据这样的方面,可以进一步改进有机无机复合体的碳物质部分的溶解性,并且可以进一步改进碳物质部分的结构控制率。另外,根据这样的方面,可以甚至更精确地控制有机无机复合体的结构。
[0297]
有机无机复合体特别优选为以下方面:其中所有碳
‑
氧键,即c
‑
o键(包括醇衍生的c
‑
o键、醚衍生的c
‑
o键和环氧衍生的c
‑
o键)和c=o键(包括羰基衍生的c=o键、羧基衍生的c=o键、酯衍生的c=o键和内酯衍生的c=o键)的总量相对于所有键,即c
‑
c键、c=c键、c
‑
h键、c
‑
o键和c=o键的总量的比例(该比例由c1s xps分析确定)落在上述范围;以及其中醚
衍生的c
‑
o键(即,c
‑
o
‑
c键)和醇衍生的c
‑
o键(即,c
‑
oh键)的总量相对于所有键,即c
‑
c键、c=c键、c
‑
h键、c
‑
o键(包括醇衍生的c
‑
o键、醚衍生的c
‑
o键和环氧衍生的c
‑
o键)和c=o键(包括羰基衍生的c=o键、羧基衍生的c=o键、酯衍生的c=o键和内酯衍生的c=o键)的总量的比例(该比例由c1s xps分析确定)落在上述范围。根据这样的方面,可以进一步改进有机无机复合体的碳物质部分的溶解性,并且可以进一步改进碳物质部分的结构控制率。另外,根据这样的方面,可以甚至更精确地控制有机无机复合体的结构。
[0298]
有机无机复合体的最优选方面为使得:所有碳
‑
氧键,即c
‑
o键(包括醇衍生的c
‑
o键、醚衍生的c
‑
o键和环氧衍生的c
‑
o键)和c=o键(包括羰基衍生的c=o键、羧基衍生的c=o键、酯衍生的c=o键和内酯衍生的c=o键)的总量相对于所有键,即c
‑
c键、c=c键、c
‑
h键、c
‑
o键和c=o键的总量的比例(该比例由c1s xps分析确定)落在上述范围;醚衍生的c
‑
o键(即,c
‑
o
‑
c键)和醇衍生的c
‑
o键(即,c
‑
oh键)的总量相对于所有碳
‑
氧键,即c
‑
o键(包括醇衍生的c
‑
o键、醚衍生的c
‑
o键和环氧衍生的c
‑
o键)和c=o键(包括羰基衍生的c=o键、羧基衍生的c=o键、酯衍生的c=o键和内酯衍生的c=o键)的总量的比例(该比例由c1s xps分析确定)落在上述范围;以及其中醚衍生的c
‑
o键(即,c
‑
o
‑
c键)和醇衍生的c
‑
o键(即,c
‑
oh键)的总量相对于所有键,即c
‑
c键、c=c键、c
‑
h键、c
‑
o键(包括醇衍生的c
‑
o键、醚衍生的c
‑
o键和环氧衍生的c
‑
o键)和c=o键(包括羰基衍生的c=o键、羧基衍生的c=o键、酯衍生的c=o键和内酯衍生的c=o键)的总量的比例(该比例由c1s xps分析确定)落在上述范围。根据这样的方面,可以进一步改进有机无机复合体的碳物质部分的溶解性,并且仍可以进一步改进碳物质部分的结构控制率。另外,根据这样的方面,仍可以甚至更精确地控制有机无机复合体的结构。
[0299]
在有机无机复合体的ir分析中在1,660cm
‑1~1,800cm
‑1范围内未观察到由c=o拉伸振动导致的峰。当在有机无机复合体的ir分析中在1,660cm
‑1~1,800cm
‑1范围内未观察到由c=o拉伸振动导致的峰时,可以更精确地控制有机无机复合体的结构。
[0300]
认为当在有机无机复合体的ir分析中在1,660cm
‑
1~1,800cm
‑
1范围内未观察到由c=o拉伸振动导致的峰时可以更精确地控制有机无机复合体的结构的原因如下所述。即,当通过使用如后所述的化合物和制造方法来制造有机无机复合体时,由于缩合反应,作为骨架的芳香环和氧官能团在反应后保留在产物中。此时,当维持芳香环的结构时(换言之,当控制结构时),醚交联的c
‑
o键或醇衍生的c
‑
o键以及氧官能团也会经历脱离并缩合而产生c
‑
c键,借此各自作为骨架的芳香环彼此结合。同时,当发生不期望的分解反应而使作为骨架的芳族结构裂解时,产生了源自于分解反应的c=o键。因此,在ir分析中,在1,660cm
‑1~1,800cm
‑1的范围内未观察到由c=o拉伸振动导致的峰的事实意味着分解反应受到抑制,因此这种有机无机复合体可以说是具有更精确控制的结构的含碳物质的材料。该范围特别优选为1,700cm
‑1~1,800cm
‑1的范围。
[0301]
当将有机无机复合体在空气气氛下且在从40℃起以10℃/min的升温条件下进行tg
‑
dta分析时,其由所得dta曲线的上升温度表示的氧化起始温度优选为200℃以上,更优选250℃以上,最优选300℃以上。当将有机无机复合体在空气气氛下且在从40℃起以10℃/min的升温条件下进行tg
‑
dta分析时,在由dta曲线的上升温度表示的氧化起始温度落在上述范围的情况下,本发明的含碳物质的材料的氧化稳定性高,即,控制了其结构,并由此维持了其骨架结构。因此,改进了其抗氧化性(抗分解性)。如果发生可能会产生c=o键的含碳
物质的材料的骨架的裂解,则骨架的稳定性会降低,从而降低抗氧化性(抗分解性)。
[0302]
如前所述,作为通过本发明的制造方法得到的含碳物质的材料的一个实施方案的有机无机复合体包含碳物质和无机物。作为通过本发明的制造方法得到的含碳物质的材料的一个实施方案的有机无机复合体包括上述的“碳物质”,并且可以典型地通过加热化合物(a)来制造,在含有化合物(a)和无机物的组合物中通过如前所述加热该组合物的加热步骤(i),该化合物(a)通过加热而引起相同分子之间和/或不同分子之间的缩合反应。可以并入作为通过本发明的制造方法得到的含碳物质的材料的一个实施方案的有机无机复合体中关于“碳物质”的以下说明作为分别关于前述<<<<1.含碳物质的材料的制造方法>>>>章节中的描述涉及的“碳物质”、<<<<2.含碳物质的材料的制造方法的典型实例>>>>中除<<2
‑
1.有机无机复合体的制造方法>>章节之外的其他章节中涉及的“碳物质”、<<<<3.含碳物质的材料>>>>章节中的描述涉及的“碳物质”、<<<<4.有机无机复合体>>>>章节中的描述涉及的“碳物质”、和<<<<5.有机无机复合体的应用>>>>章节中的描述涉及的5.有机无机复合体的“碳物质”的描述。
[0303]
碳物质中碳组分的存在可以容易地通过该材料的c1s xps分析来识别。另外,碳物质优选在其结构中具有源自苯环的蜂窝结构(石墨烯结构)。石墨烯结构的存在与否可以通过拉曼光谱分析来识别(非专利文献4)。
[0304]
在通常情况下,相对于100atm%的材料的碳原子,在碳物质中用作杂质的金属组分的总含量优选为0.1atm%以下,更优选0.01atm%以下,特别优选实质上为零。这些组分的存在可以通过荧光x射线元素分析法(xrf)对碳物质进行分析来识别。另外,当含碳物质的材料(在本章节中,有机无机复合体)通过荧光x射线元素分析法(xrf)分析时,相对于100atm%的含碳物质的材料的碳原子,用于形成含碳物质的材料(在本章节中,有机无机复合体)的除无机物的金属元素以外的金属元素的含量优选为0.1atm%以下,更优选0.01atm%以下,特别优选实质上为零。例如,在使用氧化铝作为其无机物的含碳物质的材料(在本章节中,有机无机复合体)通过荧光x射线元素分析法分析的情况下,当氧化铝中的金属元素单独为铝时,相对于100atm%的含碳物质的材料的碳原子,铝以外的金属元素的含量优选为0.1atm%以下,更优选0.01atm%以下,特别优选实质上为零。
[0305]
碳物质包含碳作为必要的构成元素,并且可以包含除碳以外的元素。这种除碳以外的元素优选为选自氧、氢、氮、硫、氟、氯、溴和碘中的至少一种元素,更优选为选自氧、氢、氮和硫中的至少一种元素,还更优选为选自氧、氢和氮中的至少一种元素,特别优选为选自氧和氢中的至少一种元素。当将在用于形成碳物质的元素中除氢以外的元素的总量设定为100atm%时,碳的量优选为60atm%以上,更优选为70atm%以上,还更优选为75atm%以上。另外,除碳以外的元素的量优选为10atm%以上。当各个元素的比率落在上述范围时,通常不溶的碳物质可以表现出令人满意的溶解性。这些元素的存在可以通过x射线光电子能谱(c1s xps)确定碳物质中元素的量来识别。另外,在含碳物质的材料(在本章节中,有机
‑
无机复合材料)的元素的量通过x射线光电子能谱(c1s xps)确定的情况下,当将在用于形成含碳物质的材料(在本章节中,有机
‑
无机复合材料)的除无机物的元素以外的元素的总量设定为100atm%,碳的量优选为60atm%以上,更优选70atm%以上,还更优选75atm%以上。另外,除碳以外的元素的量优选为10atm%以上。例如,在使用磷钨酸作为无机物的含碳物质的材料(在本章节中,有机
‑
无机复合材料)通过x射线光电子能谱(c1s xps)分析的情况
下,检测磷和钨。因此,碳的量相对于除用于形成磷钨酸的磷、钨和氧以外的元素(除氢以外)的总量(其量由磷和钨的含量计算出)的比例,以及除碳以外的元素的量相对于总量的比例优选落在上述范围。
[0306]
碳物质优选是溶剂可溶的。
[0307]
本文中,碳物质可溶于溶剂中的情况是碳物质在溶剂中的溶解度比现有技术的碳物质更优异的情况,因此可实现令人满意的可操作性。
[0308]
可以优选采用以下实施方式中的任一种作为碳物质可溶于溶剂中的方面:
[0309]
(实施方式1)碳物质的全体溶解在溶剂中的实施方式,即,碳材料仅由溶解在溶剂中的组分(组分a)形成的实施方式;和
[0310]
(实施方式2)其中碳物质的一部分溶解在溶剂中的实施方式,即,其中碳物质由溶解在溶剂中的组分(组分a)和不溶解在溶剂中的组分(组分b)形成的实施方式;在这种情况下,组分a例如是与无机物相互作用而不溶于溶剂中的部分。
[0311]
本发明中,“可溶于溶剂中”是指其中存在可溶于任何溶剂中的组分的方面。溶剂优选为例如n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺、n
‑
甲基吡咯烷酮、二甲基亚砜、丙酮、甲基乙基酮、甲基异丁基酮、四氢呋喃、甲醇、乙醇、2
‑
丙醇、丁醇、氯仿或二氯甲烷。即,优选为其中组分可溶于选自由n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺、n
‑
甲基吡咯烷酮、二甲基亚砜、丙酮、甲基乙基酮、甲基异丁基酮、四氢呋喃、甲醇、乙醇、2
‑
丙醇、丁醇、氯仿和二氯甲烷组成的组中的至少一种溶剂中的方面;更优选为其中组分可溶于选自由n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺、n
‑
甲基吡咯烷酮、二甲基亚砜和氯仿组成的组中的至少一种溶剂中的方面;还更优选为其中组分可溶于选自由n,n
‑
二甲基甲酰胺和n
‑
甲基吡咯烷酮组成的组中的至少一种溶剂中的方面;特别优选为其中存在可溶于n
‑
甲基吡咯烷酮中的组分的方面。
[0312]
碳物质可溶于溶剂中的一个实施方案例如是碳物质包含可溶于溶剂中的碳基化合物的实施方案。
[0313]
碳物质是否可溶于溶剂中可以通过例如以下判断方法来判断,该判断方法包括:将含碳物质的材料(在本章节中,有机无机复合体)混合在溶剂中,以使其浓度可以为0.001wt%,然后用超声波处理混合物1小时;使所得液体通过由ptfe制成的滤纸(孔径:0.45μm);观察已通过滤纸的液体中是否存在碳基化合物。当已通过滤纸的液体中存在碳基化合物时,该碳物质判断为包含可溶于溶剂中的碳基化合物。例如,由gl sciences inc.制造的gl chromatodisc(type 13p)可以用作由ptfe制成的滤纸。
[0314]
碳物质优选(i)在通过拉曼光谱分析得到的拉曼光谱中在g带(通常在1,550cm
‑1~1,650cm
‑1的范围内)中显示峰。因此,碳物质在通过拉曼光谱分析得到的拉曼光谱中在g带中具有峰(通常在1,550cm
‑1~1,650cm
‑1的范围内)的事实意味着碳物质具有石墨烯结构或类似于石墨烯结构的结构。当g带中的峰具有更高的强度且更窄时,可以说的是,碳物质具有更美的石墨烯结构或更美的类似于石墨烯结构的结构。
[0315]
碳物质优选(ii)在通过拉曼光谱分析得到的拉曼光谱中在d带(通常在1,300cm
‑1~1,400cm
‑1的范围内)中显示峰。具有源自于石墨烯结构的缺陷的结构的碳物质在通过拉曼光谱分析得到的拉曼光谱中在d带(通常在1,300cm
‑1~1,400cm
‑1的范围内)中显示峰。因此,碳物质在通过拉曼光谱分析得到的拉曼光谱中在d带(通常在1,300cm
‑1~1,400cm
‑1的
范围内)中具有峰的事实意味着碳物质包含官能团,并且具有源自于石墨烯结构的缺陷的结构或与源自于石墨烯结构的缺陷的结构相似的结构。当d带中的峰具有较低的强度时,可以说的是,碳物质具有更美的石墨烯结构或更美的类似于石墨烯结构的结构。另外,可以观察到d带中的峰的事实意味着通过本发明的制造方法得到的含碳物质的材料具有官能团。因此,可以改进碳物质在溶剂中的溶解性。
[0316]
碳物质优选(i)在通过拉曼光谱分析得到的拉曼光谱中在g带(通常在1,550cm
‑1~1,650cm
‑1的范围内)中显示峰,以及(ii)在通过拉曼光谱分析得到的拉曼光谱中在d带(通常在1,300cm
‑1~1,400cm
‑1的范围内)中显示峰。
[0317]
碳物质优选(iii)在通过拉曼光谱分析得到的拉曼光谱中在g
′
带(通常在2,650cm
‑1~2,750cm
‑1的范围内)中显示峰。因此,碳物质在通过拉曼光谱分析得到的拉曼光谱中在g
′
带(通常在2,650cm
‑1~2,750cm
‑1的范围内)中具有峰的事实意味着碳物质具有石墨烯结构或类似于石墨烯结构的结构。当石墨烯结构的数量为1个时,g
′
带中的峰的强度最强,并且随着石墨烯结构的层数增加,强度逐渐降低。然而,即使当强度随着石墨烯结构的层数增加而逐渐降低时,也可以观察到g
′
带中的峰。因此,g
′
带中的峰的存在可以换言之如下:碳物质具有石墨烯结构或类似于石墨烯结构的结构。g
′
带有时被称为“2d带”。
[0318]
碳物质优选(i)在通过拉曼光谱分析得到的拉曼光谱中在g带(通常在1,550cm
‑1~1,650cm
‑1的范围内)中显示峰,(ii)在通过拉曼光谱分析得到的拉曼光谱中在d带(通常在1,300cm
‑1~1,400cm
‑1的范围内)中显示峰,以及(iii)在通过拉曼光谱分析得到的拉曼光谱中在g
′
带(通常在2,650cm
‑1~2,750cm
‑1的范围内)中显示峰。
[0319]
碳物质优选(iv)在通过拉曼光谱分析得到的拉曼光谱中在d+d
′
带(通常在2,800cm
‑1~3,000cm
‑1的范围内)中显示峰。具有源自于石墨烯结构的缺陷的结构的碳物质在通过拉曼光谱分析得到的拉曼光谱中在d+d
′
带(通常在2,800cm
‑1~3,000cm
‑1的范围内)中显示峰。因此,碳物质在通过拉曼光谱分析得到的拉曼光谱中在d+d
′
带(通常在2,800cm
‑1~3,000cm
‑1的范围内)中具有峰的事实意味着碳物质包含官能团,并且具有源自于石墨烯结构的缺陷的结构或与源自于石墨烯结构的缺陷的结构相似的结构。当d+d
′
带中的峰具有较低的强度时,可以说的是,碳物质具有更美的石墨烯结构或更美的类似于石墨烯结构的结构。d+d
′
带有时称为“d+g带”。可以观察到d+d
′
带中的峰的事实也意味着通过本发明的制造方法得到的含碳物质的材料具有官能团。因而,可以改进碳物质在溶剂中的溶解性。
[0320]
碳物质优选(i)在通过拉曼光谱分析得到的拉曼光谱中在g带(通常在1,550cm
‑1~1,650cm
‑1的范围内)中显示峰,(ii)在通过拉曼光谱分析得到的拉曼光谱中在d带(通常在1,300cm
‑1~1,400cm
‑1的范围内)中显示峰,(iii)在通过拉曼光谱分析得到的拉曼光谱中在g
′
带(通常在2,650cm
‑1~2,750cm
‑1的范围内)中显示峰,以及(iv)在通过拉曼光谱分析得到的拉曼光谱中在d+d
′
带(通常在2,800cm
‑1~3,000cm
‑1的范围内)中显示峰。
[0321]
碳物质优选包含可溶于溶剂中的碳基化合物。
[0322]
作为一个实施方案,例如,碳物质(i)在通过拉曼光谱分析得到的拉曼光谱中在g带(通常在1,550cm
‑1~1,650cm
‑1的范围内)中显示峰,(ii)在通过拉曼光谱分析得到的拉曼光谱中在d带(通常在1,300cm
‑1~1,400cm
‑1的范围内)中显示峰,并且包含可溶于溶剂中的碳基化合物。
[0323]
当碳物质包含官能团,并且其石墨烯结构的一部分具有缺陷时,该缺陷可有助于
表现出碳物质在溶剂中的溶解性。
[0324]
如上所述,碳物质与常规已知的碳材料不同,具有石墨烯结构或类似于石墨烯结构的结构,因此该碳物质在溶剂中的溶解度变得更优异(例如,溶解在溶剂中的碳物质的组分的量变大,或者碳物质可以溶解的溶剂的种类数增加)。
[0325]
碳物质中的碳基化合物的分子量优选为1,000~1,300,000,更优选5,000~1,000,000,还更优选10,000~700,000,特别优选15,000~500,000,最优选20,000~300,000。碳物质中的碳基化合物的分子量落在上述范围内的情况,与特征(i)配合,使碳物质在溶剂中的溶解性更优异(例如,溶解在溶剂中的碳物质的组分的量变大,或者碳物质可以溶解的溶剂的种类数增加)。当碳物质中的碳基化合物的分子量大于1,300,000时,碳物质在溶剂中的溶解性可能劣化。当碳物质中的碳基化合物的分子量小于1,000时,作为碳物质的特征可能逐渐消失。任何这样的分子量可以通过稍后描述的途径来分析。
[0326]
碳物质中碳基化合物的含量优选为50wt%~100wt%,更优选70wt%~100wt%,还更优选90wt%~100wt%,特别优选95wt%~100wt%,最优选实质上100wt%。碳物质中碳基化合物的含量落在上述范围内的情况,与特征(i)和(ii)配合,使碳物质在溶剂中的溶解性更优异(例如,溶解在溶剂中的碳物质的组分的量变大,或者碳物质可以溶解的溶剂的种类数增加)。
[0327]
碳物质优选在通过xrd分析得到的xrd光谱图中在20
°
~30
°
范围内显示峰。即,碳物质具有石墨烯结构层叠的结构(石墨烯层叠结构)的实施方案也是一个优选的实施方案。层叠结构的存在可以使碳物质更坚固,并且可以使该物质更稳定。
[0328]
碳物质的更优选的实施方案是:其中该物质具有通过拉曼光谱分析得到的拉曼光谱的上述特征(i)~(iv)中的任一个、或其组合的实施方案;或其中该物质具有特征(i)和(ii),特征(i)、(ii)和(iii),或特征(i)、(ii)、(iii)和(iv),并且在通过xrd分析得到的xrd光谱图中在20
°
~30
°
范围内显示峰。
[0329]
碳物质可以优选以本体状态(bulk state)存在。通常,处于本体状态的物质的性质是该物质固有的性质。即,处于本体状态的物质可以确定该物质的基本性质的值,例如沸点、熔点、粘度和密度。术语“物质的物性”是指其主体部分(bulk portion)的性质。本体状态的实例包括颗粒、丸粒和膜。颗粒的存在状态例如是粉末。膜优选是自支撑膜。
[0330]
<<2
‑
2.含碳物质的颗粒的制造方法>>
[0331]
通过本发明的制造方法得到的含碳物质的材料的一个实施方案为含碳物质的颗粒。含碳物质的颗粒的典型实例包括核壳颗粒、高度碳化的核壳颗粒、中空碳细颗粒、和高度碳化的中空碳细颗粒。
[0332]
<2
‑2‑
1.核壳颗粒的制造方法>
[0333]
核壳颗粒可以通过将在加热步骤(i)之后除去通过加热化合物(a)产生的碳物质的至少一部分的碳物质除去步骤并入本发明的含碳物质的材料的制造方法中来生产。即,颗粒可以通过以下来制造:通过加热步骤(i)制造作为含碳物质的材料的一个实施方案的有机无机复合体;然后将有机无机复合体进行除去复合体中的碳物质的至少一部分的碳物质除去步骤。在碳物质除去步骤中,有机无机复合体用溶解了复合体中的碳物质的溶剂来处理。因而,可以得到核壳颗粒(核部分:无机物颗粒,壳部分:碳物质结合区域)200,其中如图2中所示,无机物颗粒20的表面被覆有碳物质结合区域(未被溶剂溶解的区域)30。这种核
壳颗粒也是含碳物质的材料。
[0334]
溶剂的实例包括n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺、n
‑
甲基吡咯烷酮、二甲基亚砜、丙酮、甲基乙基酮、甲基异丁基酮、四氢呋喃、甲醇、乙醇、2
‑
丙醇、丁醇、氯仿和二氯甲烷。其中,优选n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺、n
‑
甲基吡咯烷酮、二甲基亚砜和氯仿,更优选n,n
‑
二甲基甲酰胺和n
‑
甲基吡咯烷酮,特别优选n
‑
甲基吡咯烷酮。
[0335]
<2
‑2‑
2.高度碳化的核壳颗粒的制造方法>
[0336]
高度碳化的核壳颗粒可以通过将在加热步骤(i)和其随后的碳物质除去步骤之后进一步加热残留物的加热步骤(ii)并入本发明的含碳物质的材料的制造方法中来生产。即,进一步加热上述的核壳颗粒(核部分:无机物颗粒,壳部分:碳物质结合区域)。壳部分可以通过加热步骤(ii)来高度碳化。因而,可以得到高度碳化的核壳颗粒(核部分:无机物颗粒,壳部分:高度碳化的产物)。当将壳部分高度碳化时,可以改进所得碳物质或碳物质复合体的强度和耐热性。高度碳化的核壳颗粒也是含碳物质的材料。
[0337]
尽管加热步骤(ii)中的加热温度仅需要等于或小于核的无机组分可以承受的温度,但是具体的加热温度优选为500℃~3,000℃,更优选600℃~2,500℃,最优选700℃~2,000℃。当将加热步骤(ii)中的加热温度调节在上述范围时,可以以有效的方式使壳部分高度碳化。
[0338]
作为具体的加热时间,加热步骤(ii)中的加热时间优选为0.1小时~120小时,更优选0.5小时~100小时,还更优选1小时~50小时,最优选2小时~24小时。当将加热时间调节在上述范围时,可以以有效的方式使壳部分高度碳化。
[0339]
<2
‑2‑
3.中空碳细颗粒的制造方法>
[0340]
中空碳细颗粒可以通过将在加热步骤(i)之后除去无机物的无机物除去步骤(制造模式1)并入本发明的含碳物质的材料的制造方法中来生产。即,细颗粒可以通过以下来制造:通过加热步骤(i)制造作为含碳物质的材料的一个实施方案的有机无机复合体;然后将有机无机复合体进行除去复合体中的无机物的无机物除去步骤。
[0341]
中空碳细颗粒还可以通过将在加热步骤(i)和其随后的碳物质除去步骤之后进行除去无机物的无机物除去步骤(制造模式2)并入本发明的含碳物质的材料的制造方法中来生产。即,细颗粒可以通过以下来制造:通过加热步骤(i)和随后的碳物质除去步骤制造作为含碳物质的材料的一个实施方案的核壳颗粒;然后将核壳颗粒进行除去各颗粒中的无机物的无机物除去步骤。
[0342]
在无机物除去步骤中除去无机物的方法例如是包括用能够溶解无机物而不溶解碳物质的溶剂除去无机物的方法。具有如上所述这样的溶解特性的溶剂没有特别限制,优选为水系溶剂。如上所述优选水系溶剂的原因是,通过本发明的制造方法生产的含碳物质的材料中的碳物质通常难溶于水中,而其中的无机物通常可溶于水(特别是酸性水或碱性水)中。水系溶剂的实例包括:酸性水溶液,例如硫酸、盐酸和硝酸;和碱性水溶液,例如氢氧化钠、氢氧化钾和氨水。另外,除去步骤中的温度没有特别限制,优选为0℃~150℃,更优选20℃~100℃,因为可以有效地表现出水系溶剂的溶解特性。另外,除去步骤的物理处理没有特别限制,但是因为可以有效地表现出无机物的除去性,优选静置、搅拌、超声波处理或剪切操作,更优选搅拌、超声波处理或剪切操作。
[0343]
<2
‑2‑
4.高度碳化的中空碳细颗粒的制造方法>
[0344]
高度碳化的中空碳细颗粒可以通过将在加热步骤(i)和其随后的无机物除去步骤之后进一步加热残留物的加热步骤(ii)并入本发明的含碳物质的材料的制造方法中来制造。即,将在上述制造模式1中得到的中空碳细颗粒进一步加热。细颗粒的碳物质部分可以通过加热步骤(ii)来高度碳化。因而,可以得到高度碳化的中空碳细颗粒。当碳物质部分高度碳化时,可以改进所得的碳物质或碳物质复合体的强度和耐热性。高度碳化的中空碳细颗粒也是含碳物质的材料。
[0345]
高度碳化的中空碳细颗粒也可以通过将在加热步骤(i)、其随后的碳物质除去步骤和其随后的无机物除去步骤之后进一步加热残留物的加热步骤(ii)并入本发明的含碳物质的材料的制造方法中来制造。即,将上述的制造模式2中得到的中空碳细颗粒进一步加热。细颗粒的碳物质部分可以通过加热步骤(ii)来高度碳化。
[0346]
<<<<3.含碳物质的材料>>>>
[0347]
本发明的含碳物质的材料是包含碳物质和无机物的含碳物质的材料,并且碳物质的至少一部分和无机物的至少一部分通过共价键彼此地结合。
[0348]
在本发明的含碳物质的材料中,碳物质的至少一部分和无机物的至少一部分通过共价键彼此地结合。优选的是,碳物质的至少一部分共价地结合至无机物的最外表面的至少一部分。更优选的是,碳物质的至少一部分共价地结合至各无机物颗粒的最外表面的至少一部分。
[0349]
<<<<1.含碳物质的材料的制造方法>>>>和<<<<2.含碳物质的材料的制造方法的典型实例>>>>章节的描述可以援引至本发明的含碳物质的材料的“碳物质”和“无机物”中。
[0350]
本发明的含碳物质的材料可以通过任何适当的方法制造。本发明的含碳物质的材料可以优选地通过本发明的含碳物质的材料的制造方法来制造。
[0351]
本发明的含碳物质的材料种类繁多,并且可以采用诸如颗粒状和非颗粒状(例如,纤维状或薄膜状)等的各种形状中的任一种作为其形状。形状优选为颗粒状。
[0352]
如前述的<<<<1.含碳物质的材料的制造方法>>>>和<<<<2.含碳物质的材料的制造方法的典型实例>>>>章节中所记载的,例如,当各自采用各种形状的无机物,例如颗粒状无机物(无机物颗粒)和非颗粒状无机物(例如纤维状无机物或薄膜状无机物)作为无机物时,多种多样的含碳物质的材料可以根据各自的形状来得到。无机物优选为颗粒状无机物(无机物颗粒)。
[0353]
如前述的<<<<1.含碳物质的材料的制造方法>>>>和<<<<2.含碳物质的材料的制造方法的典型实例>>>>章节中所记载的,在使用无机物颗粒的情况下,本发明的含碳物质的材料典型地例如是包含许多无机物颗粒的块状的含碳物质的材料。在这种情况下,可以通过切碎等得到颗粒状含碳物质的材料。另外,可以通过调节化合物(a)与无机物之间的共混比例来得到颗粒状含碳物质的材料。另外,当使用纤维状无机物时,本发明的含碳物质的材料例如是纤维状的核壳颗粒或管状的中空碳物质。此外,当使用薄膜状无机物时,本发明的含碳物质的材料例如是层压的含碳物质的材料。另外,各种薄膜状碳物质可以通过进一步将这种层压的含碳物质的材料进行例如前述的加热步骤(ii)、碳物质除去步骤或无机物除去步骤来得到。
[0354]
本发明的含碳物质的材料的典型实施方案如通过<<<<2.含碳物质的材料的制造方法的典型实例>>>>章节中记载的制造方法得到的含碳物质的材料一样,例如是有机无机
复合体和含碳物质的颗粒。含碳物质的颗粒的实例包括核壳颗粒、高度碳化的核壳颗粒、中空碳细颗粒和高度碳化的中空碳细颗粒。
[0355]
本发明的含碳物质的材料在其
13
c
‑
nmr分析中优选在125ppm~135ppm的范围内显示峰。含碳物质的材料在
13
c
‑
nmr分析中在125ppm~135ppm的范围内显示峰的事实意味着含碳物质的材料包括至少含有sp2碳的碳物质。
[0356]
本发明的含碳物质的材料在
13
c
‑
nmr分析中,优选在140ppm~160ppm的范围内显示峰。含碳物质的材料在
13
c
‑
nmr分析中在140ppm~160ppm的范围内显示峰的事实意味着c
‑
o键的存在。因此,含碳物质的材料在
13
c
‑
nmr分析中,在140ppm~160ppm的范围内显示峰的事实典型地意味着含碳物质的材料在无机物的最外表面具有“具有氧的基团”(例如,具有源自于无机氧化物的氧或金属羟基的基团,并且氧和碳物质的碳彼此共价地结合。
[0357]
另外,本发明的含碳物质的材料在其
29
si
‑
nmr分析中具有源自于c
‑
o
‑
si键的峰的情况是指碳物质的至少一部分和具有si的无机物的至少一部分通过共价键而彼此结合。该情况典型地是指碳物质的至少一部分共价地结合至各二氧化硅颗粒的最外表面的至少一部分。
[0358]
<<<<2.含碳物质的材料的制造方法的典型实例>>>>章节中记载的内容可以援引至本发明的含碳物质的材料中的碳物质部分的厚度中。
[0359]
<<<<4.有机无机复合体>>>>
[0360]
本发明的有机无机复合体是包括碳物质和无机物的有机无机复合体,并且该碳物质是溶剂可溶的。
[0361]
在本发明的有机无机复合体中,<<2
‑
1.有机无机复合体的制造方法>>章节中的描述可以援引至有机无机复合体的组成和物性、以及“碳物质”中。在本发明的有机无机复合体中,<<1
‑
1.化合物(a)>>章节中的描述可以援引至“化合物(a)”中。在本发明的有机无机复合体中,<<1
‑
2.无机物>>章节中的描述可以援引至“无机物”中。
[0362]
在本发明的效果不受损的程度,本发明的有机无机复合体可以通过任何适当的方法来制造。这种制造方法典型地包括加热包含化合物(a)和无机物的组合物的加热步骤(i),该化合物(a)通过加热而引起相同分子之间和/或不同分子之间的缩合反应。包含化合物(a)和无机物的组合物中的化合物(a)可以通过加热步骤(i)加热而转变成碳物质。
[0363]
当化合物(a)的缩合反应温度为t℃时,在制造本发明的有机无机复合体时加热步骤(i)中的加热温度优选为(t
‑
150)℃以上。
[0364]
在制造本发明的有机无机复合体时的加热步骤(i)中,加热包含化合物(a)和无机物的组合物,该化合物(a)通过加热而引起相同分子之间和/或不同分子之间的缩合反应。化合物(a)与无机物之间的共混比例如下:化合物(a)的量优选为0.01wt%~1,000,000wt%,更优选0.1wt%~100,000wt%,特别优选1wt%~1,000wt%,相对于100wt%的无机物。当将化合物(a)与无机物之间的共混比例落在上述范围时,可以在更温和的条件下更简单地制造具有更精确控制的结构的有机无机复合体。无机物与化合物(a)之间的共混比例可以根据复合体的目标物性适当调节。例如,调节无机物与化合物(a)之间的共混比例能够控制要获得的有机无机复合体的物性和形态(例如,在溶剂中的溶解度,碳组分或无机组分的形状(不论该组分是颗粒状或非颗粒状),以及碳组分或无机组分的尺寸)。
[0365]
可以在本发明的效果不受损的程度将任何合适的其他组分掺入含有化合物(a)和
无机物的组合物中,该化合物(a)通过加热而引起相同分子之间和/或不同分子之间的缩合反应。这种其他组分的实例包括溶剂、催化剂、母体材料和载体。
[0366]
在制造本发明的有机无机复合体时,在加热步骤(i)中待加热的组合物仅需要在本发明的效果不受损的程度通过任何合适的方法来制备。这种方法例如是包括当组分为固态时通过任何适当的方法(例如,压碎或粉碎)将化合物(a)和无机物混合的方法。另外,该方法例如是包括通过任何适当的方法(例如,超声波处理)将化合物(a)、无机物、溶剂以及根据需要的除溶剂之外的任何其他组分混合并通过任何适当的方法(例如真空干燥)除去溶剂的方法。另外,可以根据需要进行切碎。
[0367]
当化合物(a)的缩合反应温度为t℃时,在制造本发明的有机无机复合体时加热步骤(i)中的加热温度优选为(t
‑
150)℃以上,更优选(t
‑
150)℃~(t+50)℃,还更优选(t
‑
130)℃~(t+45)℃,甚至还更优选(t
‑
100)℃~(t+40)℃,特别优选(t
‑
80)℃~(t+35)℃,最优选(t
‑
50)℃~(t+30)℃。当制造本发明的有机无机复合体时,无机物的催化能力以及无机物上的官能团与碳物质之间的反应性高,因此官能团与碳物质之间的反应可以从与如上所述的化合物(a)的缩合反应温度相比为相对较低的温度进行,以推进无机物的碳化。当将加热温度调节在上述范围时,可以在更温和的条件下更简单地制造在溶剂中具有溶解性的有机无机复合体或具有更精确控制的结构的有机无机复合体。
[0368]
化合物(a)的缩合反应温度可以通过tg
‑
dta分析确定。具体程序如下所述。
[0369]
(1)当使用一种化合物作为化合物(a)时,在氮气气氛下、且从40℃起以10℃/min的升温速度进行化合物(a)的tg
‑
dta分析,将所得的dta曲线的最低峰顶温度确定为化合物(a)的缩合反应温度(t℃)。
[0370]
(2)当使用两种以上的化合物的混合物作为化合物(a)时,在氮气气氛下、且从40℃起以10℃/min的升温速度进行混合物的tg
‑
dta分析,将所得的dta曲线的最低峰顶温度确定为化合物(a)(两种以上的化合物的混合物)的缩合反应温度(t℃)。
[0371]
(3)但是,当作为一种化合物或两种以上的化合物的混合物的化合物(a)包含例如杂质如溶剂、水分或水合水时,在某些情况下,在低于缩合反应温度的温度下观察到伴随杂质的脱离的dta峰(有时称为“杂质峰”)。在这种情况下,在忽略杂质峰的同时,确定化合物(a)的缩合反应温度。在忽略杂质峰的同时,典型地将所得的dta曲线的最低峰顶温度确定为化合物(a)的缩合反应温度。
[0372]
作为具体的加热温度,在制造本发明的有机无机复合体时加热步骤(i)中的加热温度优选为200℃~500℃,更优选220℃~400℃,还更优选230℃~350℃,最优选250℃~300℃。当将加热温度调节在上述范围时,可以在更温和的条件下更简单地制造在溶剂中具有溶解性的有机无机复合体或具有更精确控制的结构的有机无机复合体。特别地,如上所述加热步骤(i)中的加热温度低,因此有机无机复合体可以在更温和的条件下工业地制造。
[0373]
作为具体的加热时间,在制造本发明的有机无机复合体时加热步骤(i)中的加热时间优选为0.1小时~120小时,更优选0.5小时~100小时,还更优选1小时~50小时,最优选2小时~24小时。当将加热时间调节在上述范围时,可以在更温和的条件下更简单地制造在溶剂中具有溶解性的有机无机复合体或具有更精确控制的结构的有机无机复合体。
[0374]
<<<<2.含碳物质的材料的制造方法的典型实例>>>>章节中记载的内容可以援引至本发明的有机无机复合体中的碳物质部分的厚度中。
[0375]
<<<<5.有机无机复合体的应用>>>>
[0376]
本发明的有机无机复合体可以开发成各种应用。通过这种应用开发得到的典型产品是含碳物质的颗粒。含碳物质的材料种类繁多,并且可以采用诸如颗粒状和非颗粒状(例如,纤维状或薄膜状)等的各种形状中的任一种作为其形状。形状优选为颗粒状。含碳物质的颗粒的典型实例包括核
‑
壳颗粒、高度碳化的核
‑
壳颗粒、中空碳细颗粒和高度碳化的中空碳细颗粒。
[0377]
<<5
‑
1.核壳颗粒>>
[0378]
核壳颗粒可以如下来制造:在制造本发明的有机无机复合体时,在加热步骤(i)之后除去通过加热化合物(a)而生产的碳物质碳的至少一部分的物质除去步骤,然后进行除去无机物的无机物除去步骤。即,颗粒可以通过以下来制造:通过加热步骤(i)制造本发明的有机无机复合体;然后将本发明的有机无机复合体进行除去复合体中的碳物质的至少一部分的碳物质除去步骤。在碳物质除去步骤中,本发明的有机无机复合体用溶解了复合体中的碳物质的溶剂来处理。因而,可以得到核壳颗粒(核部分:无机物颗粒,壳部分:碳物质结合区域)200,其中如图2中所示,无机物颗粒20的表面被覆有碳物质结合区域(未被溶剂溶解的区域)30。
[0379]
溶剂的实例包括n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺、n
‑
甲基吡咯烷酮、二甲基亚砜、丙酮、甲基乙基酮、甲基异丁基酮、四氢呋喃、甲醇、乙醇、2
‑
丙醇、丁醇、氯仿和二氯甲烷。其中,优选n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺、n
‑
甲基吡咯烷酮、二甲基亚砜和氯仿,更优选n,n
‑
二甲基甲酰胺和n
‑
甲基吡咯烷酮,特别优选n
‑
甲基吡咯烷酮。
[0380]
<<<<2.含碳物质的材料的制造方法的典型实例>>>>章节中记载的内容可以援引至核壳颗粒中的碳物质部分的厚度中。
[0381]
<<5
‑
2.高度碳化的核壳颗粒>>
[0382]
可以如下制造高度碳化的核壳颗粒:在制造本发明的有机无机复合体时,随后在加热步骤(i)之后进行碳物质除去步骤,然后将残留物进行进一步加热残留物的加热步骤(ii)。即,进一步加热通过碳物质除去步骤得到的核壳颗粒(核部分:无机物颗粒,壳部分:碳物质结合区域)。壳部分可以通过加热步骤(ii)来高度碳化。因而,可以得到高度碳化的核壳颗粒(核部分:无机物颗粒,壳部分:高度碳化的产物)。当将壳部分高度碳化时,可以改进所得碳物质或碳物质复合体的强度和耐热性。
[0383]
尽管加热步骤(ii)中的加热温度仅需要等于或小于核的无机组分可以承受的温度,但是具体的加热温度优选为500℃~3,000℃,更优选600℃~2,500℃,最优选700℃~2,000℃。当将加热步骤(ii)中的加热温度调节在上述范围时,可以以有效的方式使壳部分高度碳化。
[0384]
作为具体的加热时间,加热步骤(ii)中的加热时间优选为0.1小时~120小时,更优选0.5小时~100小时,还更优选1小时~50小时,最优选2小时~24小时。当将加热时间调节在上述范围时,可以以有效的方式使壳部分高度碳化。
[0385]
<<<<2.含碳物质的材料的制造方法的典型实例>>>>章节中记载的内容可以援引至高度碳化的核壳颗粒中的碳物质部分的厚度中。
[0386]
<<5
‑
3.中空碳细颗粒>>
[0387]
中空碳细颗粒可以通过将在加热步骤(i)之后除去无机物的无机物除去步骤(制
造模式1)并入本发明的有机无机复合体的制造方法中来生产。即,细颗粒可以通过以下来制造:通过加热步骤(i)制造有机无机复合体;然后将有机无机复合体进行除去复合体中的无机物的无机物除去步骤。
[0388]
中空碳细颗粒还可以如下来制造(制造模式2):在制造本发明的有机无机复合体时,随后在加热步骤(i)之后进行碳物质除去步骤,然后进行除去无机物的无机物除去步骤。即,细颗粒可以通过以下来制造:通过加热步骤(i)和随后的碳物质除去步骤制造核壳颗粒;然后将核壳颗粒进行除去各颗粒中的无机物的无机物除去步骤。
[0389]
在无机物除去步骤中除去无机物的方法例如是包括用能够溶解无机物而不溶解碳物质的溶剂除去无机物的方法。具有如上所述这样的溶解特性的溶剂没有特别限制,优选为水系溶剂。如上所述优选水系溶剂的原因是,通过本发明的制造方法生产的有机无机复合体中的碳物质通常难溶于水中,而其中的无机物通常可溶于水(特别是酸性水或碱性水)中。水系溶剂的实例包括:酸性水溶液,例如硫酸、盐酸和硝酸;和碱性水溶液,例如氢氧化钠、氢氧化钾和氨水。另外,除去步骤中的温度没有特别限制,优选为0℃~150℃,更优选20℃~100℃,因为可以有效地表现出水系溶剂的溶解特性。另外,除去步骤的物理处理没有特别限制,但是因为可以有效地表现出无机物的除去性,优选静置、搅拌、超声波处理或剪切操作,更优选搅拌、超声波处理或剪切操作。
[0390]
<<<<2.含碳物质的材料的制造方法的典型实例>>>>章节中记载的内容可以援引至中空碳细颗粒中的碳物质部分的厚度中。
[0391]
<<5
‑
4.高度碳化的中空碳细颗粒>>
[0392]
可以如下制造高度碳化的中空碳细颗粒:在制造本发明的有机无机复合体时,随后在加热步骤(i)之后进行无机物除去步骤,然后将残留物进行进一步加热残留物的加热步骤(ii)。即,进一步加热在上述的制造模式1中得到的中空碳细颗粒。细颗粒的碳物质部分可以通过加热步骤(ii)来高度碳化。因而,可以得到高度碳化的中空碳细颗粒。当将碳物质部分高度碳化时,可以改进所得碳物质或碳物质复合体的强度和耐热性。
[0393]
还可以如下制造高度碳化的中空碳细颗粒:在制造本发明的有机无机复合体时,随后在加热步骤(i)之后进行碳物质除去步骤,之后进一步进行无机物除去步骤,然后进行进一步加热残留物的加热步骤(ii)。即,进一步加热在上述的制造模式2中得到的中空碳细颗粒。细颗粒的碳物质部分可以通过加热步骤(ii)来高度碳化。
[0394]
<<<<2.含碳物质的材料的制造方法的典型实例>>>>章节中记载的内容可以援引至高度碳化的中空碳细颗粒中的碳物质部分的厚度中。
[0395]
实施例
[0396]
以下通过实施例具体描述本发明,但是本发明不限于这些实施例。除非另有规定,术语“份”是指“重量份”,符号“%”是指“wt%”。另外,本文所用的术语“重量”可以用术语“质量”代替;条件是本文中有关c1s xps的部分中的符号“%”是指“atm%”。
[0397]
<拉曼光谱分析>
[0398]
在以下条件下用以下装置进行拉曼光谱分析。
[0399]
测量装置:显微拉曼光谱仪(nrs
‑
3100,由jasco corporation制造)
[0400]
测量条件:使用波长为532nm的激光,物镜的放大倍率为20,ccd捕获时间为1秒,扫描次数为64次(分辨率=4cm
‑1)。
[0401]
在拉曼分析中,可能会出现g
′
带和d+d
′
带,同时还彼此重叠,因此,可以将d+d
′
带分析为特别是具有肩部的宽峰。在这种情况下,肩峰的拐点视为是g
′
带的峰。
[0402]
<xrd分析>
[0403]
xrd测量用全自动水平x射线衍射仪(由rigaku corporation,smart lab制造)在以下条件下进行。
[0404]
cukα1
‑
射线:0.15406nm
[0405]
扫描范围:10
°
~90
°
[0406]
x射线输出设置:45kv
‑
200ma
[0407]
步长:0.020
°
[0408]
扫描速度:0.5
°
min
‑1~4
°
min
‑1[0409]
通过在将样品装入手套箱中的密闭样品台中而保持惰性气氛的状态下进行xrd测量。
[0410]
<c1s xps分析>
[0411]
用光电子能谱仪(axis
‑
ultra,由shimadzu corporation制造),在以下条件下进行c1s xps测量。
[0412]
来源:mg(双节点)
[0413]
发射:10ma
[0414]
阳极:10kv
[0415]
分析仪:通过能量(pass energy):40
[0416]
测量范围:c1s:296ev~270ev
[0417]
扫描次数:10次
[0418]
分析条件:在以下能量下,对于各官能团分离源自于c1s轨道的峰,并从各自的面积计算出峰的比率。在以下五个能量下分离出源自于典型种类的官能团的峰:(1)在288.3ev处分离出源自于
‑
coo
‑
、内酯和一些酮的峰;(2)在286.2ev处分离出源自于c=o和环氧基的峰;(3)在285.6ev处分离出源自于c
‑
oh和c
‑
o
‑
c的峰;(4)在284.3ev处分离出源自于六元环c=c的峰;(5)在283.6ev处分离出源自于c
‑
c、c
‑
h和五元环c=c的峰。然而,为了方便比率计算,将峰(4)和(5)的面积共同地计算。与c1s xps有关的部分中的符号“%”是指“atm%”。在表1中,与项目(1)、(2)和(3)相对应的峰的比率分别显示为(a)、(b)和(c),并且对应于项目(4)和(5)的峰的总比率显示为(d)。
[0419]
<tg
‑
dta分析>
[0420]
用以下装置在以下条件下进行tg
‑
dta分析。
[0421]
测量装置:同步热重分析仪(由seiko instruments inc.制造,tg/dta6200)
[0422]
如下所述确定化合物(a)的缩合反应温度。
[0423]
(1)当使用一种化合物作为化合物(a)时,在氮气气氛下、且从40℃起以10℃/min的升温速度进行化合物(a)的tg
‑
dta分析,将所得的dta曲线的最低峰顶温度确定为化合物(a)的缩合反应温度(t℃)。
[0424]
(2)当使用两种以上的化合物的混合物作为化合物(a)时,在氮气气氛下、且从40℃起以10℃/min的升温速度进行混合物的tg
‑
dta分析,将所得的dta曲线的最低峰顶温度确定为化合物(a)(两种以上的化合物的混合物)的缩合反应温度(t℃)。
[0425]
(3)但是,当作为一种化合物或两种以上的化合物的混合物的化合物(a)包含例如杂质如溶剂、水分或水合水时,在某些情况下,在低于缩合反应温度的温度下观察到伴随杂质的脱离的dta峰(有时称为“杂质峰”)。在这种情况下,在忽略杂质峰的同时,确定化合物(a)的缩合反应温度。具体地,在忽略杂质峰的同时,将所得的dta曲线的最低峰顶温度确定为化合物(a)的缩合反应温度。
[0426]
有机无机复合体的氧化起始温度通过以下来评估:在空气气氛下从40℃起以10℃/min的升温速度将复合体进行tg
‑
dta分析;从所得的dta曲线中在dta曲线的上升温度中最低的上升温度来评估氧化起始温度。
[0427]
<ir分析>
[0428]
用以下装置在以下条件下进行ft
‑
ir分析。
[0429]
测量装置:傅立叶变换红外光谱仪(ft/ir
‑
4200,由jasco corporation制造)
[0430]
测量条件:通过具有mct检测器的漫反射傅里叶变换光谱(drift)在分辨率4cm
‑1和扫描次数128次下进行测量。
[0431]
样品条件:使用通过将样品和kbr以1:50的重量比混合而得到的混合物。
[0432]
<
13
c
‑
nmr测量和
29
si
‑
nmr测量>
[0433]
用以下装置在以下条件下进行
13
c
‑
nmr测量和
29
si
‑
nmr测量。
[0434]
装置:bruker avance neo 400mhz/263ghz 9.4t,dnp系统
[0435]
探头:2ch 3.2mm dnp探头
[0436]
样品管:3.2mm蓝宝石转子+特氟龙嵌件+氧化锆盖
[0437]
测量温度:105k~107k
[0438]
魔角旋转(mas):10khz
[0439]
脉冲程序:cp(1h
‑
13
c、1h
‑
29
si)
[0440]
cp接触时间:3ms
[0441]
样品制备条件:40mg样品+20μl tekpol/1,1,2,2
‑
四氯乙烷(16mm)
[0442]
<分子量的测量>
[0443]
如下所述测量含碳物质的材料中的碳物质的分子量。将含碳物质的材料混合在n,n
‑
二甲基甲酰胺(含有0.1%的libr)中,以使各碳物质的含量变为0.02wt%,并且将混合物用超声波处理1小时。之后,使混合物通过由ptfe(0.45μm)制成的滤纸来进行预处理,然后将滤液通过使用n,n
‑
二甲基甲酰胺(含有0.1%的libr)作为展开剂来进行凝胶渗透色谱(gpc,hlc
‑
8220gpc,由tosoh corporation制造),然后以聚苯乙烯换算来计算分子量。碳物质中的最大分子量由其峰的上升点算出。
[0444]
<热传导特性(热扩散率和热传导率)的测量>
[0445]
关于用作热传导特性的热扩散率,使用由ai
‑
phase co.,ltd.制造的m3 2型,测量膜在厚度方向上的热扩散率。用作热传导特性的热传导率由表述“膜的比热
×
密度
×
热扩散率”算出。
[0446]
<sps烧结>
[0447]
在以下条件下用以下装置进行sps烧结。
[0448]
装置:labox
‑
125c,由sinterland,inc.制造
[0449]
气氛:真空下
[0450]
温度升高:10℃/min(从~5pa起升温)
[0451]
烧结温度:610℃
[0452]
保持时间:5分钟
[0453]
加压:50mpa(从升温到烧结)
[0454]
冷却:无压、自然冷却
[0455]
<粉末电阻的测量>
[0456]
如下所述测量粉末电阻。4mm厚的特氟龙板打出1厘米的方孔,并且将粉末装入该孔中。将粉末从其两侧夹在铜电极之间,并测量粉末的电阻。使用的装置是由tektronix,inc.制造的高电阻计6517bj,并且在10v的施加电压下测量电阻。
[0457]
[实施例1]:间苯三酚+二氧化硅颗粒+250℃
×
1小时
[0458]
将间苯三酚(由tokyo chemical industry co.,ltd.制造,熔点:220℃,缩合反应温度:330℃,理论比表面积:750m2/g)与二氧化硅颗粒(由fuji silysia chemical ltd.制造,产品名:"q
‑
10ht60315",比表面积:259m2/g)充分地混合,以使间苯三酚层的厚度为二氧化硅颗粒层的厚度的1.5倍大。
[0459]
将所得的混合物在真空中密封在石英安瓿管中,然后在预先加热至250℃的电炉中加热1小时。
[0460]
因而,得到包括碳物质(1a)和无机氧化物(1b)的有机无机复合体(1)。
[0461]
将有机无机复合体(1)分散于n
‑
甲基吡咯烷酮(nmp)中,并且其碳组分的溶解性通过前述中所述的方式而确认。结果,该组分可溶于溶剂中。
[0462]
所得的有机无机复合体(1)的c1s xps分析的结果显示在图3(xps光谱(c1s))和表1中。从表1中发现,有机无机复合体(1)为包括碳物质和无机氧化物的有机无机复合体。所有碳
‑
氧键(c
‑
o键和c=o键)的总量与所有碳键(c
‑
c键、c=c键、c
‑
h键、c
‑
o键、和c=o键)的总量的比例为26%,醚衍生的c
‑
o键和醇衍生的c
‑
o键的总量与所有碳
‑
氧键(c
‑
o键和c=o键)的总量的比例为62%,醚衍生的c
‑
o键和醇衍生的c
‑
o键的总量与所有碳键(c
‑
c键、c=c键、c
‑
h键、c
‑
o键、和c=o键)的总量的比例为16%。从前述中发现了复合体具有高的结构控制率。
[0463]
当将间苯三酚在氮气气氛、以10℃/min的升温条件进行tg
‑
dta分析时,其在温度500℃的重量m500与其在温度50℃的初始重量m50的重量比(m500/m50)为0.49。从前述中发现了,间苯三酚即使在碳化后也可以充分存在于无机氧化物上。
[0464]
所得的有机无机复合体(1)的tg
‑
dta分析中的dta分析的结果显示在图4中。根据图4,当有机无机复合体(1)在空气气氛下且在从40℃起以10℃/min的升温条件下进行tg
‑
dta分析时,其由所得dta曲线的上升温度表示的氧化起始温度为200℃。从前述中发现了复合体具有高的耐氧化性。
[0465]
此外,通过对所得的有机无机复合体(1)进行ir分析而得到的结果显示在图5中。根据图5,在有机无机复合体(1)的ir光谱中,未观察到源自于c=o结构的在1,660cm
‑1~1,800cm
‑1范围内的峰,因此可以高度控制复合体的结构。
[0466]
此外,所得有机无机复合体(1)的拉曼光谱显示在图6中。拉曼光谱在1,365cm
‑1、1,590cm
‑1、2,650cm
‑1、和2,835cm
‑1处具有峰,因此发现碳物质(1a)是具有石墨烯结构并且具有其中石墨烯结构层叠的结构的、含有碳基化合物的碳物质。
[0467]
如上所述,根据本发明的制造方法,可以在温和条件下简单地制造可溶于溶剂中且具有精确控制的结构的含碳物质的材料。另外,如上所述,根据本发明,可以提供可溶于溶剂中、具有精确控制的结构、并且可以在温和条件下工业上制造的有机无机复合体。
[0468]
[实施例2]:间苯三酚+氧化铝颗粒+250℃
×
1小时
[0469]
除了将二氧化硅颗粒变更为氧化铝颗粒(由一般社团法人“日本催化学会”免费分发的样品“jrc
‑
alo7”,比表面积:180m2/g)以外,以与实施例1相同的方式得到包括碳物质(2a)和无机氧化物(2b)的有机无机复合体(2)。
[0470]
将有机无机复合体(2)分散于n
‑
甲基吡咯烷酮(nmp)中,并且其碳组分的溶解性通过前述中所述的方式而确认。结果,该组分可溶于溶剂中。
[0471]
所得的有机无机复合体(2)的c1s xps分析的结果显示在图3(xps光谱(c1s))和表1中。从表1中发现,有机无机复合体(2)为包括碳物质和无机氧化物的有机无机复合体。所有碳
‑
氧键(c
‑
o键和c=o键)的总量与所有碳键(c
‑
c键、c=c键、c
‑
h键、c
‑
o键、和c=o键)的总量的比例为29%,醚衍生的c
‑
o键和醇衍生的c
‑
o键的总量与所有碳
‑
氧键(c
‑
o键和c=o键)的总量的比例为59%,醚衍生的c
‑
o键和醇衍生的c
‑
o键的总量与所有碳键(c
‑
c键、c=c键、c
‑
h键、c
‑
o键、和c=o键)的总量的比例为17%。从前述中发现了复合体具有高的结构控制率。
[0472]
所得的有机无机复合体(2)的tg
‑
dta分析中的dta分析的结果显示在图4中。根据图4,当有机无机复合体(2)在空气气氛下且在从40℃起以10℃/min的升温条件下进行tg
‑
dta分析时,其由所得dta曲线的上升温度表示的氧化起始温度为230℃。从前述中发现了复合体具有高的耐氧化性。
[0473]
此外,通过对所得的有机无机复合体(2)进行ir分析而得到的结果显示在图5中。根据图5,在有机无机复合体(2)的ir光谱中,存在源自于c=o结构的在1,660cm
‑1~1,800cm
‑1范围内的峰,因此复合体具有由c1s xps确定的高的结构控制率,但是具有由ft
‑
ir分析确定的稍微低的结构控制率。
[0474]
此外,所得有机无机复合体(2)的拉曼光谱显示在图6中。拉曼光谱在1,340cm
‑1、1,585cm
‑1、2,650cm
‑1、和2,835cm
‑1处具有峰,因此发现碳物质(2a)是具有石墨烯结构并且具有其中石墨烯结构层叠的结构的、含有碳基化合物的碳物质。
[0475]
如上所述,根据本发明的制造方法,可以在温和条件下简单地制造可溶于溶剂中且具有精确控制的结构的含碳物质的材料。另外,如上所述,根据本发明,可以提供可溶于溶剂中、具有精确控制的结构、并且可以在温和条件下工业上制造的有机无机复合体。
[0476]
[实施例3]:间苯三酚+二氧化钛颗粒+250℃
×
1小时
[0477]
除了将二氧化硅颗粒变更为二氧化钛颗粒(由一般社团法人“日本催化学会”免费分发的样品“jrc
‑
tio
‑
4(2)”,比表面积:50m2/g)以外,以与实施例1相同的方式得到包括碳物质(3a)和无机氧化物(3b)的有机无机复合体(3)。
[0478]
将有机无机复合体(3)分散于n
‑
甲基吡咯烷酮(nmp)中,并且其碳组分的溶解性通过前述中所述的方式而确认。结果,该组分可溶于溶剂中。
[0479]
所得的有机无机复合体(3)的c1s xps分析的结果显示在图3(xps光谱(c1s))和表1中。从表1中发现,有机无机复合体(3)为包括碳物质和无机氧化物的有机无机复合体。所有碳
‑
氧键(c
‑
o键和c=o键)的总量与所有碳键(c
‑
c键、c=c键、c
‑
h键、c
‑
o键、和c=o键)的
总量的比例为27%,醚衍生的c
‑
o键和醇衍生的c
‑
o键的总量与所有碳
‑
氧键(c
‑
o键和c=o键)的总量的比例为56%,醚衍生的c
‑
o键和醇衍生的c
‑
o键的总量与所有碳键(c
‑
c键、c=c键、c
‑
h键、c
‑
o键、和c=o键)的总量的比例为15%。从前述中发现了复合体具有高的结构控制率。
[0480]
所得的有机无机复合体(3)的tg
‑
dta分析中的dta分析的结果显示在图4中。根据图4,当有机无机复合体(3)在空气气氛下且在从40℃起以10℃/min的升温条件下进行tg
‑
dta分析时,其由所得dta曲线的上升温度表示的氧化起始温度为200℃。从前述中发现了复合体具有高的耐氧化性。
[0481]
此外,通过对所得的有机无机复合体(3)进行ir分析而得到的结果显示在图5中。根据图5,在有机无机复合体(3)的ir光谱中,未观察到源自于c=o结构的在1,660cm
‑1~1,800cm
‑1范围内的峰,因此复合体具有由c1s xps确定的高的结构控制率,但是具有由ft
‑
ir分析确定的稍微低的结构控制率。
[0482]
此外,所得有机无机复合体(3)的拉曼光谱显示在图6中。拉曼光谱在1,340cm
‑1、1,580cm
‑1、2,650cm
‑1、和2,820cm
‑1处具有峰,因此发现碳物质(3a)是具有石墨烯结构并且具有其中石墨烯结构层叠的结构的、含有碳基化合物的碳物质。
[0483]
如上所述,根据本发明的制造方法,可以在温和条件下简单地制造可溶于溶剂中且具有精确控制的结构的含碳物质的材料。另外,如上所述,根据本发明,可以提供可溶于溶剂中、具有精确控制的结构、并且可以在温和条件下工业上制造的有机无机复合体。
[0484]
[实施例4]:间苯三酚+杂多酸颗粒(hpw)+250℃
×
1小时
[0485]
除了将二氧化硅颗粒变更为用作杂多酸颗粒的hpw(由fujifilm wako pure chemical corporation制造,12
‑
钨(vi)磷酸n
‑
水合物,比表面积:278m2/g)以外,以与实施例1相同的方式得到包括碳物质(4a)和无机氧化物(4b)的有机无机复合体(4)。
[0486]
将有机无机复合体(4)分散于n
‑
甲基吡咯烷酮(nmp)中,并且其碳组分的溶解性通过前述中所述的方式而确认。结果,该组分可溶于溶剂中。
[0487]
所得的有机无机复合体(4)的c1s xps分析的结果显示在图3(xps光谱(c1s))和表1中。从表1中发现,有机无机复合体(4)为包括碳物质和无机氧化物的有机无机复合体。所有碳
‑
氧键(c
‑
o键和c=o键)的总量与所有碳键(c
‑
c键、c=c键、c
‑
h键、c
‑
o键、和c=o键)的总量的比例为29%,醚衍生的c
‑
o键和醇衍生的c
‑
o键的总量与所有碳
‑
氧键(c
‑
o键和c=o键)的总量的比例为76%,醚衍生的c
‑
o键和醇衍生的c
‑
o键的总量与所有碳键(c
‑
c键、c=c键、c
‑
h键、c
‑
o键、和c=o键)的总量的比例为22%。从前述中发现了复合体具有高的结构控制率。
[0488]
所得的有机无机复合体(4)的tg
‑
dta分析中的dta分析的结果显示在图4中。根据图4,当有机无机复合体(4)在空气气氛下且在从40℃起以10℃/min的升温条件下进行tg
‑
dta分析时,其由所得dta曲线的上升温度表示的氧化起始温度为300℃。从前述中发现了复合体具有高的耐氧化性。
[0489]
此外,通过对所得的有机无机复合体(4)进行ir分析而得到的结果显示在图5中。根据图5,在有机无机复合体(4)的ir光谱中,未观察到源自于c=o结构的在1,660cm
‑1~1,800cm
‑1范围内的峰,因此可以高度控制复合体的结构。
[0490]
此外,有机无机复合体(4)的拉曼光谱显示在图6中。拉曼光谱在1,350cm
‑1、1,
585cm
‑1、2,650cm
‑1、和2,810cm
‑1处具有峰,因此发现碳物质(4a)是具有石墨烯结构并且具有其中石墨烯结构层叠的结构的、含有碳基化合物的碳物质。另外,测量碳物质(4a)部分的分子量。结果,该部分的重均分子量为8,000,且最大分子量为50,000。
[0491]
如上所述,根据本发明的制造方法,可以在温和条件下简单地制造可溶于溶剂中且具有精确控制的结构的含碳物质的材料。另外,如上所述,根据本发明,可以提供可溶于溶剂中、具有精确控制的结构、并且可以在温和条件下工业上制造的有机无机复合体。
[0492]
[实施例5]:间苯三酚+杂多酸颗粒(hpmo)+250℃
×
1小时
[0493]
除了将二氧化硅颗粒变更为用作杂多酸颗粒的hpmo(由fujifilm wako pure chemical corporation制造,12
‑
钼(vi)磷酸n
‑
水合物)以外,以与实施例1相同的方式得到包括碳物质(5a)和无机氧化物(5b)的有机无机复合体(5)。
[0494]
将有机无机复合体(5)分散于n
‑
甲基吡咯烷酮(nmp)中,并且其碳组分的溶解性通过前述中所述的方式而确认。结果,该组分可溶于溶剂中。
[0495]
所得的有机无机复合体(5)的c1s xps分析的结果显示在图7(xps光谱(c1s))和表1中。从表1中发现,有机无机复合体(5)为包括碳物质和无机氧化物的有机无机复合体。所有碳
‑
氧键(c
‑
o键和c=o键)的总量与所有碳键(c
‑
c键、c=c键、c
‑
h键、c
‑
o键、和c=o键)的总量的比例为27%,醚衍生的c
‑
o键和醇衍生的c
‑
o键的总量与所有碳
‑
氧键(c
‑
o键和c=o键)的总量的比例为63%,醚衍生的c
‑
o键和醇衍生的c
‑
o键的总量与所有碳键(c
‑
c键、c=c键、c
‑
h键、c
‑
o键、和c=o键)的总量的比例为17%。从前述中发现了复合体具有高的结构控制率。
[0496]
此外,通过对所得的有机无机复合体(5)进行ir分析而得到的结果显示在图8中。根据图8,在有机无机复合体(5)的ir光谱中,未观察到源自于c=o结构的在1,660cm
‑1~1,800cm
‑1范围内的峰,因此可以高度控制复合体的结构。
[0497]
如上所述,根据本发明的制造方法,可以在温和条件下简单地制造可溶于溶剂中且具有精确控制的结构的含碳物质的材料。另外,如上所述,根据本发明,可以提供可溶于溶剂中、具有精确控制的结构、并且可以在温和条件下工业上制造的有机无机复合体。
[0498]
[实施例6]:间苯三酚+杂多酸颗粒(hsiw)+250℃
×
1小时
[0499]
除了将二氧化硅颗粒变更为用作杂多酸颗粒的hsiw(由japan new metals co.,ltd.制造,硅钨酸)以外,以与实施例1相同的方式得到包括碳物质(6a)和无机氧化物(6b)的有机无机复合体(6)。
[0500]
将有机无机复合体(6)分散于n
‑
甲基吡咯烷酮(nmp)中,并且其碳组分的溶解性通过前述中所述的方式而确认。结果,该组分可溶于溶剂中。
[0501]
所得的有机无机复合体(6)的c1s xps分析的结果显示在图7(xps光谱(c1s))和表1中。从表1中发现,有机无机复合体(6)为包括碳物质和无机氧化物的有机无机复合体。所有碳
‑
氧键(c
‑
o键和c=o键)的总量与所有碳键(c
‑
c键、c=c键、c
‑
h键、c
‑
o键、和c=o键)的总量的比例为29%,醚衍生的c
‑
o键和醇衍生的c
‑
o键的总量与所有碳
‑
氧键(c
‑
o键和c=o键)的总量的比例为72%,醚衍生的c
‑
o键和醇衍生的c
‑
o键的总量与所有碳键(c
‑
c键、c=c键、c
‑
h键、c
‑
o键、和c=o键)的总量的比例为21%。从前述中发现了复合体具有高的结构控制率。
[0502]
此外,通过对所得的有机无机复合体(6)进行ir分析而得到的结果显示在图8中。
根据图8,在有机无机复合体(6)的ir光谱中,未观察到源自于c=o结构的在1,660cm
‑1~1,800cm
‑1范围内的峰,因此可以高度控制复合体的结构。
[0503]
如上所述,根据本发明的制造方法,可以在温和条件下简单地制造可溶于溶剂中且具有精确控制的结构的含碳物质的材料。另外,如上所述,根据本发明,可以提供可溶于溶剂中、具有精确控制的结构、并且可以在温和条件下工业上制造的有机无机复合体。
[0504]
[实施例7]:间苯三酚+杂多酸颗粒(hpvmo)+250℃
×
1小时
[0505]
除了将二氧化硅颗粒变更为用作杂多酸颗粒的hpvmo(由japan new metals co.,ltd.制造,磷钒钼酸)以外,以与实施例1相同的方式得到包括碳物质(7a)和无机氧化物(7b)的有机无机复合体(7)。
[0506]
将有机无机复合体(7)分散于n
‑
甲基吡咯烷酮(nmp)中,并且其碳组分的溶解性通过前述中所述的方式而确认。结果,该组分可溶于溶剂中。
[0507]
所得的有机无机复合体(7)的c1s xps分析的结果显示在图7(xps光谱(c1s))和表1中。从表1中发现,有机无机复合体(7)为包括碳物质和无机氧化物的有机无机复合体。所有碳
‑
氧键(c
‑
o键和c=o键)的总量与所有碳键(c
‑
c键、c=c键、c
‑
h键、c
‑
o键、和c=o键)的总量的比例为29%,醚衍生的c
‑
o键和醇衍生的c
‑
o键的总量与所有碳
‑
氧键(c
‑
o键和c=o键)的总量的比例为55%,醚衍生的c
‑
o键和醇衍生的c
‑
o键的总量与所有碳键(c
‑
c键、c=c键、c
‑
h键、c
‑
o键、和c=o键)的总量的比例为16%。从前述中发现了复合体具有高的结构控制率。
[0508]
此外,通过对所得的有机无机复合体(7)进行ir分析而得到的结果显示在图8中。根据图8,在有机无机复合体(7)的ir光谱中,未观察到源自于c=o结构的在1,660cm
‑1~1,800cm
‑1范围内的峰,因此可以高度控制复合体的结构。
[0509]
如上所述,根据本发明的制造方法,可以在温和条件下简单地制造可溶于溶剂中且具有精确控制的结构的含碳物质的材料。另外,如上所述,根据本发明,可以提供可溶于溶剂中、具有精确控制的结构、并且可以在温和条件下工业上制造的有机无机复合体。
[0510]
[实施例8]:间苯三酚+杂多酸颗粒(hpwmo)+250℃
×
1小时
[0511]
除了将二氧化硅颗粒变更为用作杂多酸颗粒的hpwmo(由japan new metals co.,ltd.制造,磷钨钼酸)以外,以与实施例1相同的方式得到包括碳物质(8a)和无机氧化物(8b)的有机无机复合体(8)。
[0512]
将有机无机复合体(8)分散于n
‑
甲基吡咯烷酮(nmp)中,并且其碳组分的溶解性通过前述中所述的方式而确认。结果,该组分可溶于溶剂中。
[0513]
所得的有机无机复合体(8)的c1s xps分析的结果显示在图7(xps光谱(c1s))和表1中。从表1中发现,有机无机复合体(8)为包括碳物质和无机氧化物的有机无机复合体。所有碳
‑
氧键(c
‑
o键和c=o键)的总量与所有碳键(c
‑
c键、c=c键、c
‑
h键、c
‑
o键、和c=o键)的总量的比例为27%,醚衍生的c
‑
o键和醇衍生的c
‑
o键的总量与所有碳
‑
氧键(c
‑
o键和c=o键)的总量的比例为70%,醚衍生的c
‑
o键和醇衍生的c
‑
o键的总量与所有碳键(c
‑
c键、c=c键、c
‑
h键、c
‑
o键、和c=o键)的总量的比例为19%。从前述中发现了复合体具有高的结构控制率。
[0514]
此外,通过对所得的有机无机复合体(8)进行ir分析而得到的结果显示在图8中。根据图8,在有机无机复合体(8)的ir光谱中,未观察到源自于c=o结构的在1,660cm
‑1~1,
800cm
‑1范围内的峰,因此可以高度控制复合体的结构。
[0515]
如上所述,根据本发明的制造方法,可以在温和条件下简单地制造可溶于溶剂中且具有精确控制的结构的含碳物质的材料。另外,如上所述,根据本发明,可以提供可溶于溶剂中、具有精确控制的结构、并且可以在温和条件下工业上制造的有机无机复合体。
[0516]
[实施例9]:间苯三酚+二氧化硅颗粒+300℃
×
3小时,中空碳细颗粒
[0517]
将300mg间苯三酚(由tokyo chemical industry co.,ltd.制造,熔点:220℃,缩合反应温度:330℃)溶解在3g异丙醇中,并且将1,000mg二氧化硅球形细颗粒(由nippon shokubai co.,ltd.制造,平均粒径:0.19μm)加入溶液中,然后通过超声波处理充分混合内容物。
[0518]
异丙醇通过常温真空干燥从所得混合物中除去,并将残留的块状产物切碎,然后在300℃下加热3小时。
[0519]
因而,得到包括碳物质(9a)和无机氧化物(9b)的有机无机复合体(9)。
[0520]
有机无机复合体(9)的拉曼光谱显示在图9中。拉曼光谱在1,375cm
‑1、1,600cm
‑1、2,700cm
‑1、和2,890cm
‑1处具有峰,因此发现碳物质(9a)是具有石墨烯结构并且具有其中石墨烯结构层叠的结构的、含有碳基化合物的碳物质。另外,测量碳物质(9a)部分的分子量。结果,该部分的重均分子量为200,000,且最大分子量为1,100,000。
[0521]
将所得的有机无机复合体(9)用n
‑
甲基吡咯烷酮(nmp)处理。因而,除去碳物质(9a),由此得到各自在无机氧化物(9b)的表面被碳物质结合区域被覆的核壳颗粒(9)(核部分:无机氧化物颗粒,壳部分:碳物质结合区域)。
[0522]
将核壳颗粒(9)进一步在700℃下煅烧1小时。因而,将各个壳部分中的碳物质高度碳化,因此得到高度碳化的核壳颗粒(9)。高度碳化的核壳颗粒(9)的sem照片显示在图10中,并且高度碳化的核壳颗粒(9)的表面的拉曼光谱显示在图11中。从图10和图11中发现,高度碳化的核壳颗粒(9)的表面(壳部分)各自包括高度碳化的碳物质,而不损害其形状。
[0523]
高度碳化的核壳颗粒(9)的
13
c
‑
nmr分析和
29
si
‑
nmr分析的结果分别显示在图12和图13中。首先,在
13
c
‑
nmr中,能够观察到在129ppm处的峰和在140ppm~160ppm范围内的肩峰。认为这些峰是源自于sp2碳的存在(129ppm),以及无机物表面上的氧原子与源自于碳物质的碳原子之间的键(140ppm~160ppm)。在后述的比较例3中,推测的是没有形成sp2碳,并且主要形成sp3碳,并且该sp3碳隐藏在溶剂峰的后面。另外,在
29
si
‑
nmr中,与用作原料的二氧化硅球形细颗粒和后述的比较例3相比,能够清楚地观察到源自于si
‑
o
‑
c键的峰。因此,发现在源自于碳物质的碳原子与无机物的最外表面上的原子之间形成共价键。如上所述,碳原子仅共价地结合至无机物的最外表面。因此,该峰的强度小,由此能够观察到作为肩峰的该峰。
[0524]
测量高度碳化的核壳颗粒(9)的粉末电阻为3
×
105ω
·
cm。用作原料的二氧化硅球形细颗粒的粉末电阻为10
14
ω
·
cm量级,因此发现当细颗粒转变成核壳颗粒时,它们的导电性能够约109倍地改进。
[0525]
高度碳化的核壳颗粒(9)的碳部分的厚度由其tg
‑
dta分析来评估。评估的方法包括:在空气气氛下以10℃/min将用作原料的二氧化硅球形细颗粒和高度碳化的核壳颗粒(9)各自的温度从室温升高到1,000℃,以使核壳颗粒的碳组分燃烧;计算用作原料的二氧化硅球形细颗粒与高度碳化的核壳颗粒(9)在燃烧后的重量之差,以分析碳组分的量。根据
该方法,高度碳化的核壳颗粒(9)的碳组分的含量为1.39wt%。当将各二氧化硅球形细颗粒的直径设定为0.19μm,并且将碳组分的密度设定为2时,计算出碳组分的平均厚度为0.49nm。
[0526]
将500mg核壳颗粒(9)在10%氢氧化钠水溶液中进行超声波洗涤5小时,过滤以提供20mg的中空碳细颗粒(9)。考虑到滤液是无色透明的和除去产物的量,仅碳组分残留,并且能够除去各核壳颗粒中的无机组分。可能是由于前述原因,实现了各自具有中空结构的中空碳细颗粒(9)的形成。
[0527]
[实施例10]:间苯三酚+铜颗粒+300℃
×
2小时,高度碳化的核壳颗粒
[0528]
将1g间苯三酚(由tokyo chemical industry co.,ltd.制造,熔点:220℃,缩合反应温度:330℃)溶解在200g丙酮中,并且将20g铜颗粒(由ecka granules germany gmbh制造,粒径:95vol%以上的颗粒各自的粒径为36μm以下)加入溶液中,然后通过超声波处理充分混合内容物。
[0529]
丙酮通过常温真空干燥从所得混合物中除去,并将残留的块状产物切碎,然后在300℃下加热2小时。
[0530]
因而,得到包括碳物质(10a)和无机氧化物(10b)的有机无机复合体(10)。
[0531]
将所得的有机无机复合体(10)用n,n
‑
二甲基甲酰胺(dmf)处理。因而,除去碳物质(10a),因此得到各自在无机氧化物(10b)的表面被碳物质结合区域被覆的核壳颗粒(10)(核部分:无机氧化物颗粒,壳部分:碳物质结合区域)。
[0532]
将核壳颗粒(10)进一步在700℃下煅烧1小时。因而,将各个壳部分中的碳物质高度碳化,因此得到高度碳化的核壳颗粒(10)。高度碳化的核壳颗粒(10)的表面的拉曼光谱显示在图14中。从图14中发现,高度碳化的核壳颗粒(10)的表面(壳部分)各自包括高度碳化的碳物质,而不损害其形状。
[0533]
[实施例11]:间苯三酚+铝颗粒+300℃
×
2小时,高度碳化的核壳颗粒
[0534]
将0.2g间苯三酚(由tokyo chemical industry co.,ltd.制造,熔点:220℃,缩合反应温度:330℃)溶解在200g丙酮中,并且将2g铝颗粒(由ecka granules germany gmbh制造,d50=5μm)加入溶液中。内容物通过超声波处理充分混合,并且干燥而提供铝颗粒
‑
间苯三酚混合体。将混合体用kugelrohr在氮气气氛、300℃下煅烧2小时。因而,得到包括碳物质(11a)和无机氧化物(11b)的有机无机复合体(11)。
[0535]
将所得的有机无机复合体(11)在n,n
‑
二甲基甲酰胺(dmf)中进行超声波处理,并且将处理后的产物通过离心分离除去过量的碳物质(11a)来纯化。因而,得到各自在无机氧化物(11b)的表面被碳物质结合区域被覆的核壳颗粒(11)(核部分:无机氧化物颗粒,壳部分:碳物质结合区域)。
[0536]
将核壳颗粒(11)进一步在600℃下煅烧2小时。因而,将各个壳部分中的碳物质高度碳化,因此得到高度碳化的核壳颗粒(11)。高度碳化的核壳颗粒(11)的表面的拉曼光谱显示在图15中。从图15中发现,高度碳化的核壳颗粒(11)的表面(壳部分)各自包括高度碳化的碳物质,而不损害其形状。
[0537]
另外,将所得的高度碳化的核壳颗粒(11)进行sps烧结以提供铝
‑
碳合金烧结体。作为比较,将用作原料的铝颗粒也进行sps烧结以生产铝烧结体。测定各烧结体的维氏硬度。结果,得到了如图16所示的图,因此发现在使用通过本发明中的镀碳技术生产的核壳颗
粒的情况下,在烧结时能够改进颗粒的强度(硬度)。即,通过本发明的制造方法得到的含碳物质的材料可以应用于对烧结体赋予强度的用途(烧结体强度改进剂)。
[0538]
[实施例12]:间苯三酚+钛酸钡颗粒+300℃
×
2小时,高度碳化的核壳颗粒
[0539]
将0.2g间苯三酚(由tokyo chemical industry co.,ltd.制造,熔点:220℃,缩合反应温度:330℃)溶解在200g丙酮中,并且将2g钛酸钡颗粒(由fujifilm wako pure chemical corporation制造)加入溶液中。内容物通过超声波处理充分混合,并且干燥而提供钛酸钡颗粒
‑
间苯三酚混合体。将混合体用kugelrohr在氮气气氛、300℃下煅烧2小时。因而,得到包括碳物质(12a)和无机氧化物(12b)的有机无机复合体(12)。
[0540]
将所得的有机无机复合体(12)在n,n
‑
二甲基甲酰胺(dmf)中进行超声波处理,并且将处理后的产物通过离心分离除去过量的碳物质(12a)来纯化。因而,得到各自在无机氧化物(12b)的表面被碳物质结合区域被覆的核壳颗粒(12)(核部分:无机氧化物颗粒,壳部分:碳物质结合区域)。
[0541]
将核壳颗粒(12)进一步在700℃下煅烧2小时。因而,将各个壳部分中的碳物质高度碳化,因此得到高度碳化的核壳颗粒(12)。高度碳化的核壳颗粒(12)的表面的拉曼光谱显示在图17中。从图17中发现,高度碳化的核壳颗粒(12)的表面(壳部分)各自包括高度碳化的碳物质,而不损害其形状。
[0542]
[实施例13]:间苯三酚+钛酸锶颗粒+300℃
×
2小时,高度碳化的核壳颗粒
[0543]
将0.2g间苯三酚(由tokyo chemical industry co.,ltd.制造,熔点:220℃,缩合反应温度:330℃)溶解在200g丙酮中,并且将2g钛酸锶颗粒(由sigma
‑
aldrich,inc.制造)加入溶液中。内容物通过超声波处理充分混合,并且干燥而提供钛酸锶颗粒
‑
间苯三酚混合体。将混合体用kugelrohr在氮气气氛、300℃下煅烧2小时。因而,得到包括碳物质(13a)和无机氧化物(13b)的有机无机复合体(13)。
[0544]
将所得的有机无机复合体(13)在n,n
‑
二甲基甲酰胺(dmf)中进行超声波处理,并且将处理后的产物通过离心分离除去过量的碳物质(13a)来纯化。因而,得到各自在无机氧化物(13b)的表面被碳物质结合区域被覆的核壳颗粒(13)(核部分:无机氧化物颗粒,壳部分:碳物质结合区域)。
[0545]
将核壳颗粒(13)进一步在700℃下煅烧2小时。因而,将各个壳部分中的碳物质高度碳化,因此得到高度碳化的核壳颗粒(13)。高度碳化的核壳颗粒(13)的表面的拉曼光谱显示在图18中。从图18中发现,高度碳化的核壳颗粒(13)的表面(壳部分)各自包括高度碳化的碳物质,而不损害其形状。
[0546]
[实施例14]:间苯三酚+铌酸锂颗粒+300℃
×
2小时,高度碳化的核壳颗粒
[0547]
将0.2g间苯三酚(由tokyo chemical industry co.,ltd.制造,熔点:220℃,缩合反应温度:330℃)溶解在200g丙酮中,并且将2g铌酸锂颗粒(由sigma
‑
aldrich,inc.制造)加入溶液中。内容物通过超声波处理充分混合,并且干燥而提供铌酸锂颗粒
‑
间苯三酚混合体。将混合体用kugelrohr在氮气气氛、300℃下煅烧2小时。因而,得到包括碳物质(14a)和无机氧化物(14b)的有机无机复合体(14)。
[0548]
将所得的有机无机复合体(14)在n,n
‑
二甲基甲酰胺(dmf)中进行超声波处理,并且将处理后的产物通过离心分离除去过量的碳物质(14a)来纯化。因而,得到各自在无机氧化物(14b)的表面被碳物质结合区域被覆的核壳颗粒(14)(核部分:无机氧化物颗粒,壳部
分:碳物质结合区域)。
[0549]
将核壳颗粒(14)进一步在700℃下煅烧2小时。因而,将各个壳部分中的碳物质高度碳化,因此得到高度碳化的核壳颗粒(14)。高度碳化的核壳颗粒(14)的表面的拉曼光谱显示在图19中。从图19中发现,高度碳化的核壳颗粒(14)的表面(壳部分)各自包括高度碳化的碳物质,而不损害其形状。
[0550]
[实施例15]:间苯三酚+硅颗粒+300℃
×
2小时,高度碳化的核壳颗粒
[0551]
将0.2g间苯三酚(由tokyo chemical industry co.,ltd.制造,熔点:220℃,缩合反应温度:330℃)溶解在200g丙酮中,并且将2g硅颗粒(由yy company制造)加入溶液中。内容物通过超声波处理充分混合,并且干燥而提供硅颗粒
‑
间苯三酚混合体。将混合体用kugelrohr在氮气气氛、300℃下煅烧2小时。因而,得到包括碳物质(15a)和无机氧化物(15b)的有机无机复合体(15)。
[0552]
将所得的有机无机复合体(15)在n,n
‑
二甲基甲酰胺(dmf)中进行超声波处理,并且将处理后的产物通过离心分离除去过量的碳物质(15a)来纯化。因而,得到各自在无机氧化物(15b)的表面被碳物质结合区域被覆的核壳颗粒(15)(核部分:无机氧化物颗粒,壳部分:碳物质结合区域)。
[0553]
将核壳颗粒(15)进一步在700℃下煅烧2小时。因而,将各个壳部分中的碳物质高度碳化,因此得到高度碳化的核壳颗粒(15)。高度碳化的核壳颗粒(15)的表面的拉曼光谱显示在图20中。从图20中发现,高度碳化的核壳颗粒(15)的表面(壳部分)各自包括高度碳化的碳物质,而不损害其形状。
[0554]
[实施例16]:间苯三酚+氮化硼颗粒+300℃
×
2小时,高度碳化的核壳颗粒
[0555]
将0.2g间苯三酚(由tokyo chemical industry co.,ltd.制造,熔点:220℃,缩合反应温度:330℃)溶解在200g丙酮中,并且将2g氮化硼颗粒(由showa denko k.k.制造)加入溶液中。内容物通过超声波处理充分混合,并且干燥而提供氮化硼颗粒
‑
间苯三酚混合体。将混合体用kugelrohr在氮气气氛、300℃下煅烧2小时。因而,得到包括碳物质(16a)和无机氧化物(16b)的有机无机复合体(16)。
[0556]
将所得的有机无机复合体(16)在n,n
‑
二甲基甲酰胺(dmf)中进行超声波处理,并且将处理后的产物通过离心分离除去过量的碳物质(16a)来纯化。因而,得到各自在无机氧化物(16b)的表面被碳物质结合区域被覆的核壳颗粒(16)(核部分:无机氧化物颗粒,壳部分:碳物质结合区域)。
[0557]
将核壳颗粒(16)进一步在700℃下煅烧2小时。因而,将各个壳部分中的碳物质高度碳化,因此得到高度碳化的核壳颗粒(16)。高度碳化的核壳颗粒(16)的表面的拉曼光谱显示在图21中。从图21中发现,高度碳化的核壳颗粒(16)的表面(壳部分)各自包括高度碳化的碳物质,而不损害其形状。
[0558]
[实施例17]:间苯三酚+氧化铝颗粒+300℃
×
2小时,高度碳化的核壳颗粒s
[0559]
将0.2g间苯三酚(由tokyo chemical industry co.,ltd.制造,熔点:220℃,缩合反应温度:330℃)溶解在200g丙酮中,并且将2g氧化铝颗粒(由showa denko k.k.制造)加入溶液中。内容物通过超声波处理充分混合,并且干燥而提供氧化铝颗粒
‑
间苯三酚混合体。将混合体用kugelrohr在氮气气氛、300℃下煅烧2小时。因而,得到包括碳物质(17a)和无机氧化物(17b)的有机无机复合体(17)。
[0560]
将所得的有机无机复合体(17)在n,n
‑
二甲基甲酰胺(dmf)中进行超声波处理,并且将处理后的产物通过离心分离除去过量的碳物质(17a)来纯化。因而,得到各自在无机氧化物(17b)的表面被碳物质结合区域被覆的核壳颗粒(17)(核部分:无机氧化物颗粒,壳部分:碳物质结合区域)。
[0561]
将核壳颗粒(17)进一步在700℃下煅烧2小时。因而,将各个壳部分中的碳物质高度碳化,因此得到高度碳化的核壳颗粒(17)。高度碳化的核壳颗粒(17)的表面的拉曼光谱显示在图22中。从图22中发现,高度碳化的核壳颗粒(17)的表面(壳部分)各自包括高度碳化的碳物质,而不损害其形状。
[0562]
另外,使用所得的高度碳化的核壳颗粒(17)和用作原料的氧化铝颗粒,如下提供混合物“聚甲基丙烯酸甲酯(pmma)/氧化铝”和混合物“pmma/高度碳化的核壳颗粒(17)”:将pmma和用作原料的氧化铝颗粒在dmf中混合,以使“pmma:氧化铝颗粒”的体积比变为1:2,然后干燥以提供前一种混合物;将pmma和高度碳化的核壳颗粒(17)在dmf中混合,以使“pmma:高度碳化的核壳颗粒(17)”的体积比变为1:2,然后干燥以提供后一种混合物。分析了各个混合物在其厚度方向上的热传导特性。结果发现,如表2所示,使用高度碳化的核壳颗粒(17)实现了导热性的改进。各个混合物的比热和密度由混合比来计算,并且此处使用的值分别为920j/kg
·
k和3,000kg/m3。即,本发明的有机无机复合体可以应用于热传导特性改进(热传导特性改进剂)的用途。
[0563]
[实施例18]:间苯三酚+氧化镁颗粒+300℃
×
2小时,高度碳化的核壳颗粒
[0564]
将0.2g间苯三酚(由tokyo chemical industry co.,ltd.制造,熔点:220℃,缩合反应温度:330℃)溶解在200g丙酮中,并且将2g氧化镁颗粒(mgo,由ube industries,ltd.制造)加入溶液中。内容物通过超声波处理充分混合,并且干燥而提供氧化镁颗粒
‑
间苯三酚混合体。将混合体用kugelrohr在氮气气氛、300℃下煅烧2小时。因而,得到包括碳物质(18a)和无机氧化物(18b)的有机无机复合体(18)。
[0565]
将所得的有机无机复合体(18)在n,n
‑
二甲基甲酰胺(dmf)中进行超声波处理,并且将处理后的产物通过离心分离除去过量的碳物质(18a)来纯化。因而,得到各自在无机氧化物(18b)的表面被碳物质结合区域被覆的核壳颗粒(18)(核部分:无机氧化物颗粒,壳部分:碳物质结合区域)。
[0566]
将核壳颗粒(18)进一步在700℃下煅烧2小时。因而,将各个壳部分中的碳物质高度碳化,因此得到高度碳化的核壳颗粒(18)。高度碳化的核壳颗粒(18)的表面的拉曼光谱显示在图23中。从图23中发现,高度碳化的核壳颗粒(18)的表面(壳部分)各自包括高度碳化的碳物质,而不损害其形状。
[0567]
[实施例19]:间苯三酚+氮化铝颗粒+300℃
×
2小时,高度碳化的核壳颗粒
[0568]
将0.2g间苯三酚(由tokyo chemical industry co.,ltd.制造,熔点:220℃,缩合反应温度:330℃)溶解在200g丙酮中,并且将2g氮化铝颗粒(由tokuyama corporation制造)加入溶液中。内容物通过超声波处理充分混合,并且干燥而提供氮化铝颗粒
‑
间苯三酚混合体。将混合体用kugelrohr在氮气气氛、300℃下煅烧2小时。因而,得到包括碳物质(19a)和无机氧化物(19b)的有机无机复合体(19)。
[0569]
将所得的有机无机复合体(19)在n,n
‑
二甲基甲酰胺(dmf)中进行超声波处理,并且将处理后的产物通过离心分离除去过量的碳物质(19a)来纯化。因而,得到各自在无机氧
化物(19b)的表面被碳物质结合区域被覆的核壳颗粒(19)(核部分:无机氧化物颗粒,壳部分:碳物质结合区域)。
[0570]
将核壳颗粒(19)进一步在700℃下煅烧2小时。因而,将各个壳部分中的碳物质高度碳化,因此得到高度碳化的核壳颗粒(19)。高度碳化的核壳颗粒(19)的表面的拉曼光谱显示在图24中。从图24中发现,高度碳化的核壳颗粒(19)的表面(壳部分)各自包括高度碳化的碳物质,而不损害其形状。
[0571]
[实施例20]:间苯三酚+磷酸锂铁颗粒+300℃
×
2小时,高度碳化的核壳颗粒
[0572]
将0.2g间苯三酚(由tokyo chemical industry co.,ltd.制造,熔点:220℃,缩合反应温度:330℃)溶解在200g丙酮中,并且将2g磷酸锂铁颗粒(由toshima manufacturing co.,ltd.制造)加入溶液中。内容物通过超声波处理充分混合,并且干燥而提供磷酸锂铁颗粒
‑
间苯三酚混合体。将混合体用kugelrohr在氮气气氛、300℃下煅烧2小时。因而,得到包括碳物质(20a)和无机氧化物(20b)的有机无机复合体(20)。
[0573]
将所得的有机无机复合体(20)在n,n
‑
二甲基甲酰胺(dmf)中进行超声波处理,并且将处理后的产物通过离心分离除去过量的碳物质(20a)来纯化。因而,得到各自在无机氧化物(20b)的表面被碳物质结合区域被覆的核壳颗粒(20)(核部分:无机氧化物颗粒,壳部分:碳物质结合区域)。
[0574]
将核壳颗粒(20)进一步在700℃下煅烧2小时。因而,将各个壳部分中的碳物质高度碳化,因此得到高度碳化的核壳颗粒(20)。高度碳化的核壳颗粒(20)的表面的拉曼光谱显示在图25中。从图25中发现,高度碳化的核壳颗粒(20)的表面(壳部分)各自包括高度碳化的碳物质,而不损害其形状。
[0575]
[实施例21]:间苯三酚+二氧化硅颗粒+250℃
×
2小时,高度碳化的核壳颗粒,不同溶剂
[0576]
将1g间苯三酚(由tokyo chemical industry co.,ltd.制造,熔点:220℃,缩合反应温度:330℃)溶解在30g丙酮中,并且将10g二氧化硅球形细颗粒(由nippon shokubai co.,ltd.制造,平均粒径:0.19μm)加入溶液中,然后通过超声波处理充分混合内容物。
[0577]
丙酮通过常温真空干燥从所得混合物中除去,并将残留的块状产物切碎,然后在250℃下加热2小时。
[0578]
因而,得到包括碳物质(21a)和无机氧化物(21b)的有机无机复合体(21)。
[0579]
有机无机复合体(21)的拉曼光谱显示在图26中。发现碳物质(21a)是具有石墨烯结构并且具有其中石墨烯结构层叠的结构的、含有碳基化合物的碳物质。
[0580]
将所得的有机无机复合体(21)在丙酮中进行超声波处理,并且将处理后的产物通过离心分离除去过量的碳物质(21a)来纯化。因而,得到各自在无机氧化物(21b)的表面被碳物质结合区域被覆的核壳颗粒(21)(核部分:无机氧化物颗粒,壳部分:碳物质结合区域)。
[0581]
将核壳颗粒(21)进一步在700℃下煅烧1小时。因而,将各个壳部分中的碳物质高度碳化,因此得到高度碳化的核壳颗粒(21)。高度碳化的核壳颗粒(21)的表面的拉曼光谱显示在图27中。
[0582]
[实施例22]:间苯三酚+氮化硼+250℃
×
2小时,高度碳化的核壳颗粒,不同溶剂
[0583]
除了使用氮化硼作为原料以外,以与实施例21相同的方式分别得到有机无机复合
体(22)、核壳颗粒(22)和高度碳化的核壳颗粒(22)。有机无机复合体(22)和高度碳化的核壳颗粒(22)的拉曼光谱分别显示在图28和图29中。从实施例21和22可以看出,能够通过诸如煅烧温度调节等的操作来改变所使用的溶剂。
[0584]
[实施例23]:六羟基三亚苯+二氧化硅颗粒+350℃
×
2小时,高度碳化的核壳颗粒,不同原料
[0585]
将1g 2,3,6,7,10,11
‑
六羟基三亚苯(由tokyo chemical industry co.,ltd.制造,熔点:无,缩合反应温度:430℃)溶解在30g dmf中,并且将10g二氧化硅球形细颗粒(由nippon shokubai co.,ltd.制造,平均粒径:0.19μm)加入溶液中,然后通过超声波处理充分混合内容物。
[0586]
dmp利用蒸发器从所得混合物中除去,并将残留的块状产物切碎,然后在350℃下加热2小时。
[0587]
因而,得到包括碳物质(23a)和无机氧化物(23b)的有机无机复合体(23)。
[0588]
有机无机复合体(23)的拉曼光谱显示在图30中。拉曼光谱在1,340cm
‑1、1,590cm
‑1、2,700cm
‑1、和2,895cm
‑1处具有峰,因此发现碳物质(23a)是具有石墨烯结构并且具有其中石墨烯结构层叠的结构的、含有碳基化合物的碳物质。
[0589]
将所得的有机无机复合体(23)在dmf中进行超声波处理,并且将处理后的产物通过离心分离除去过量的碳物质(23a)来纯化。因而,得到各自在无机氧化物(23b)的表面被碳物质结合区域被覆的核壳颗粒(23)(核部分:无机氧化物颗粒,壳部分:碳物质结合区域)。
[0590]
将核壳颗粒(23)进一步在700℃下煅烧1小时。因而,将各个壳部分中的碳物质高度碳化,因此得到高度碳化的核壳颗粒(23)。高度碳化的核壳颗粒(23)的表面的拉曼光谱显示在图31中。
[0591]
[实施例24]:(+)
‑
儿茶酚+二氧化硅颗粒+250℃
×
2小时,高度碳化的核壳颗粒,不同原料
[0592]
将1g(+)
‑
儿茶酚水合物(由tokyo chemical industry co.,ltd.制造,熔点:无,缩合反应温度:270℃)溶解在30g丙酮中,并且将10g二氧化硅球形细颗粒(由nippon shokubai co.,ltd.制造,平均粒径:0.19μm)加入溶液中,然后通过超声波处理充分混合内容物。儿茶酚是水合物,因此,tg
‑
dta中的缩合反应温度通过以下来确定:忽略脱水峰;且确定儿茶酚在脱水后进行缩合的温度。
[0593]
丙酮通过常温真空干燥从所得混合物中除去,并将残留的块状产物切碎,然后在250℃下加热2小时。
[0594]
因而,得到包括碳物质(24a)和无机氧化物(24b)的有机无机复合体(24)。
[0595]
有机无机复合体(24)的拉曼光谱显示在图32中。拉曼光谱在13cm
‑1、15cm
‑1、27cm
‑1和28cm
‑1处具有峰,因此发现碳物质(24a)是具有石墨烯结构并且具有其中石墨烯结构层叠的结构的、含有碳基化合物的碳物质。
[0596]
将所得的有机无机复合体(24)在dmf中进行超声波处理,并且将处理后的产物通过离心分离除去过量的碳物质(24a)来纯化。因而,得到各自在无机氧化物(24b)的表面被碳物质结合区域被覆的核壳颗粒(24)(核部分:无机氧化物颗粒,壳部分:碳物质结合区域)。
[0597]
将核壳颗粒(24)进一步在700℃下煅烧1小时。因而,将各个壳部分中的碳物质高度碳化,因此得到高度碳化的核壳颗粒(24)。高度碳化的核壳颗粒(24)的表面的拉曼光谱显示在图33中。
[0598]
[比较例1]
[0599]
除了将加热温度设定为170℃以外,通过实施例9所述的方法得到有机无机复合体(c1)。然而,通过拉曼分析没有观察到复合体的碳物质的信号,并且仅观察到源自于其低分子量化合物的荧光。即,发现间苯三酚未充分碳化。
[0600]
[比较例2]
[0601]
除了将加热温度设定为270℃以外,通过实施例23所述的方法得到有机无机复合体(c2)。然而,通过拉曼分析没有观察到复合体的碳物质的信号,并且仅观察到源自于其低分子量化合物的荧光。即,发现六羟基三亚苯未充分碳化。
[0602]
[比较例3]
[0603]
将实施例9中使用的二氧化硅球形细颗粒在氮气气氛、700℃下焙烧2小时。经焙烧的颗粒通过使用快速碳涂覆仪(由sanyu electron co.,ltd.制造,sc
‑
701ct)在气相中用碳被覆,以生产出颗粒。从该颗粒的拉曼分析中识别出碳的存在。该颗粒的
13
c
‑
nmr分析和
29
si
‑
nmr分析的结果分别示于图12和图13中。作为与实施例9的比较结果,发现了在比较例3中得到的各颗粒的表面上都存在碳,但不是sp2碳,并且不存在si
‑
o
‑
c键,因此该碳不是通过共价键而存在于各颗粒的表面上(当然,在用作原料的二氧化硅球形细颗粒中也无法观察到该键)。当在与实施例比较时考虑比较例3的结果时,发现了根据本发明,能够提供其无机物的表面通过共价键被sp2碳组分牢固被覆的这种含碳物质的材料。
[0604]
[参考例1]
[0605]
除了将加热温度设定为520℃以外,通过实施例9中记载的方法得到有机无机复合物(r1)。然而,复合体的碳组分不溶于n
‑
甲基吡咯烷酮(nmp),因此无法进行后续处理。
[0606]
从上述实施例等中发现了,根据本发明的制造方法,可溶于溶剂中的含碳物质的材料或具有精确控制的结构的含碳物质的材料能够在温和的条件下简单地制造。还发现了,根据本发明,能够提供这种含碳物质的材料。还发现了,使用本发明的其碳物质可溶于溶剂中的有机无机复合体能够相对容易地实现核壳型复合体或中空碳物质。
[0607][0608]
表2
[0609][0610]
产业上的可利用性
[0611]
通过本发明的制造方法得到的含碳物质的材料和本发明的含碳物质的材料可以各自有效地用作例如碳被覆无机颗粒和中空碳细颗粒的工业生产时的材料,该碳被覆无机颗粒和中空碳细颗粒各自可用作例如轻质、优异润滑性、优异导电性、优异导热性和优异抗氧化性的填料。假定使用这些材料的应用包括:固体润滑剂;添加到润滑油等中的润滑用添加剂;以及添加到无机材料或诸如树脂等的有机材料中的导电助剂、抗静电剂、强度赋予剂、摩擦减少剂或导热性赋予剂。另外,本发明的有机无机复合体可以有效地用作例如碳被覆无机颗粒和中空碳细颗粒的工业生产时的材料,该碳被覆无机颗粒和中空碳细颗粒各自可用作例如轻质、优异润滑性、优异导电性、优异导热性和优异抗氧化性的填料。假定使用这些材料的应用包括:固体润滑剂;添加到润滑油等中的润滑用添加剂;以及添加到无机材料或诸如树脂等的有机材料中的导电助剂、抗静电剂、强度赋予剂、摩擦减少剂或导热性赋予剂。
[0612]
附图标记说明
[0613]
10 碳物质
[0614]
20 无机物颗粒
[0615]
30 碳物质结合区域
[0616]
100 有机无机复合体
[0617]
200 核壳颗粒
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1