一种酮咯酸氨丁三醇中间体化合物的制作方法
1.本发明属于药物合成技术领域,具体涉及一种酮咯酸氨丁三醇中间体化合物。
背景技术:
2.酮咯酸氨丁三醇(ketorolac tromethamine),化学名为(
±
)-5-苯甲酰基-2,3-二氢-1h
-ꢀ
吡咯里嗪-1-甲酸氨丁三醇盐,是一种非甾体消炎药,由美国syntex公司开发上市,具有强力止痛及中度抗炎和解热作用,可口服或肌注,用于缓解中度至剧烈的术后疼痛。酮咯酸氨丁三醇通过抑制前列腺素的合成来发挥镇痛、抗炎和解热作用,30mg的止痛作用相当于12mg吗啡,但无成瘾性。其化学结构如下所示:
[0003][0004]
目前报道的关于酮咯酸氨丁三醇的合成路线有很多,如王华等在文献《镇痛药酮咯酸合成工艺的改进》(中国药物化学杂志,2002,12(4):228-229)报道了以吡咯为起始原料,经苯甲酰化、n-烃化、c-烃化反应,再进行环合得5-苯甲酰-2,3-二氢-1h-吡咯里嗪-1,1-二甲酸乙酯、再水解反应制得酮咯酸,反应式如下:
[0005][0006]
上述文献中合成其关键二酯基中间体采用过量的高锰酸钾和醋酸锰,使用含锰的金属盐后,产生的工艺废水处理困难,产品颜色深,后处理需要采用活性炭或者硅胶多步脱色纯化,从而又产生废活性炭和废硅胶等固废问题难以解决。
[0007]
为了解决上述合成二酯基中间体采用过量的高锰酸钾和醋酸锰问题,中国专利申请 cn108191876报道了使用副物为水的双氧水作为氧化剂,且使用铁盐为催化剂,取代了大量的锰盐。
[0008]
鉴于现有技术存在的较多问题,研究寻找一条反应条件温和,操作过程简便,产品收率高、纯度高,生产成本低的适合工业化生产酮咯酸氨丁三醇的中间体制备方法仍是目前需要解决的问题。
技术实现要素:
[0009]
针对目前酮咯酸氨丁三醇制备技术存在的问题,本发明提供了一种新的酮咯酸氨丁三醇中间体化合物及其制备方法,及利用该新中间体制备酮咯酸氨丁三醇中间体的新方
法。该方法反应条件温和,操作过程简便,生产成本低,所制得的目标产品具有较高的纯度、收率。
[0010]
本发明的具体技术方案如下:
[0011]
一种酮咯酸氨丁三醇中间体化合物,结构如下所示:
[0012][0013]
一种酮咯酸氨丁三醇中间体化合物ⅳ的制备方法,包括如下步骤:将化合物ii加入有机溶剂中,控温滴加氯化亚砜,滴加完毕,恒温反应,tlc检测,反应完毕,过滤,浓缩,烘干得化合物ⅳ,其合成路线如下:
[0014][0015]
优选的,所述有机溶剂为四氢呋喃、乙腈、乙醚、甲基叔丁基醚中的一种或其组合;其中特别优选四氢呋喃。
[0016]
优选的,所述化合物ii与氯化亚砜的投料摩尔比为1:1.2~2.0;其中特别优选1:1.4。
[0017]
优选的,所述反应温度为10~35℃,特别优选20~25℃。
[0018]
所述化合物ⅳ用于制备酮咯酸氨丁三醇中间体的用途。
[0019]
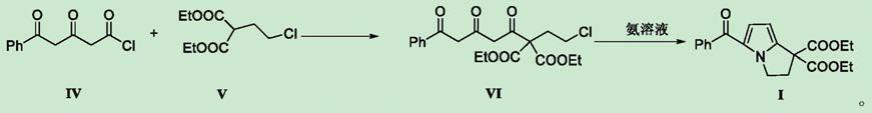
所述化合物ⅳ用于制备酮咯酸氨丁三醇中间体化合物i的用途,制备方法包括如下步骤:化合物iv与化合v在碱的作用下反应得化合物vi,化合物vi再与氨溶液反应,得化合物i,反应式如下:
[0020][0021]
优选的,在以下部分进一步详细的描述以上步骤:
[0022]
化合物vi的制备
[0023]
将化合物v加入到反应溶剂中,搅拌,控温分批缓慢加入碱,加入完毕,恒温反应;反应完毕,降温,将化合物ⅳ滴加到反应体系,滴加完毕,控温反应,反应结束,调节ph;减压蒸除溶剂,残余物加入萃取溶剂萃取,收集有机层,过滤,干燥,得化合物vi。
[0024]
优选的,所述化合物v与化合物iv的投料摩尔比为1:1.2~2.0;其中特别优选1:1.3。
[0025]
优选的,所述碱为氢化钠、氢化锂、二异丙基氨基锂、双(三甲基硅基)氨基锂、双(三甲基硅基)氨基钠、双(三甲基硅烷基)氨基钾中的一种或其组合。
[0026]
优选的,所述化合物v与碱的的投料摩尔比为1:1.5~2.5;其中特别优选1:2.0。
[0027]
优选的,所述加入碱的控温温度为-20~-5℃;其中特别优选-10℃。
[0028]
优选的,所述滴加化合物ⅳ的控温温度为-30~-10℃;其中特别优选-20℃。
[0029]
优选的,所述反应溶剂为四氢呋喃、乙腈、乙醚、甲基叔丁基醚中的一种或其组合;
其中特别优选四氢呋喃。
[0030]
优选的,所述ph值为5~7。
[0031]
优选的,所述萃取溶剂为二氯甲烷、三氯甲烷、乙酸乙酯、甲基叔丁基醚中的一种或其组合,其中特别优选二氯甲烷。
[0032]
化合物i的制备
[0033]
将化合物vi及氨溶液和反应溶剂加入高压釜,氨气保护,控温反应。反应结束,冷却至室温,减压蒸除溶剂,加入萃取溶剂和水萃取,收集有机层,无水硫酸钠干燥,过滤,旋蒸,干燥得化合物i;
[0034]
优选的,所述化合物vi与氨的投料摩尔比为1:2.0~5.0;其中特别优选1:4.0。
[0035]
优选的,所述氨的溶液为甲醇氨溶液或乙醇氨溶液;其中特别优选甲醇氨溶液。
[0036]
优选的,所述反应溶剂为甲醇、乙醇、四氢呋喃、乙腈、甲苯中的一种或其组合;其中特别优选甲醇。
[0037]
优选的,所述反应温度为50~80℃;其中特别优选60℃。
[0038]
优选的,所述萃取溶剂为二氯甲烷、三氯甲烷、乙酸乙酯中的一种或其组合;其中特别优选二氯甲烷。
[0039]
与现有技术相比,本发明取得的技术效果是:
[0040]
(1)提供了一种新的酮咯酸氨丁三醇中间体化合物iv,提供了利用该中间体合成酮咯酸氨丁三醇中间体化合物的新方法,该新中间体在合成酮咯酸氨丁三醇过程中操作简便;
[0041]
(2)新方法避免使用了剧毒和强腐蚀性的物质,反应步骤少,操作简单,适合工业化生产;
[0042]
(3)提高了反应收率和纯度,降低了成本。
具体实施方式
[0043]
下面通过实施例来进一步说明本发明,应该正确理解的是:本发明的实施例仅仅是用于说明本发明,而不是对本发明的限制,所以,在本发明的方法前提下对本发明的简单改进均属于本发明要求保护的范围。
[0044]
实验所用物料:化合物可购买,也可参照现有公开的技术制备;其他实验所用物料未标明来源和规格的均为市售分析纯或化学纯。
[0045]
以下各实施例中,未详细描述的各种过程和方法是本领域中公知的常规方法。
[0046]
化合物iv结构表征:
[0047][0048]
化合物iv的高分辨质谱:esi-hrms:m/z=225.0301[m+h]
+1
;h nmr(400mhz,dmso-d6) δ:7.92-7.98(m,2h),7.57-7.48(m,3h),4.24(s,2h),3.92(s,2h).
13
c nmr(101mhz, dmso-d6)δ:201.0,197.5,170.5,136.0,132.4,129.0,128.7,62.5,56.0.
[0049]
化合物vi结构表征:
[0050][0051]
化合物vi的高分辨质谱:esi-hrms:m/z=441.1300[m+h]
+1
;1h nmr(400mhz,dmso-d6) δ:7.99-8.02(m,2h),7.46-7.55(m,3h),4.26(q,j=4.8hz,4h),3.74(t,j=3.6hz,2h), 2.92-2.96(m,2h),2.85-2.88(m,2h),2.38(t,j=3.6hz,2h),1.23(t,j=4.8hz,6h)。
13
c nmr (101mhz,dmso-d6)δ:198.9,197.5,193.9,168.2,136.0,132.4,129.0,128.7,68.9,62.1,61.3, 56.1,40.8,31.1,14.3.
[0052]
化合物iv的制备
[0053]
实施例1
[0054]
将化合物ii(10.3g,0.05mol)加入四氢呋喃(50ml)中,控温20~25℃,搅拌10min,滴加化合物iii(8.33g,0.07mol),滴加完毕,继续保温搅拌1h,tcl检测反应,反应完毕,过滤,浓缩,烘干得化合物ⅳ,收率98.5%,hplc纯度99.94%。
[0055]
实施例2
[0056]
将化合物ii(10.3g,0.05mol)加入乙腈(50ml)中,控温10~15℃,搅拌10min,滴加化合物iii(7.14g,0.06mol),滴加完毕,继续保温搅拌1h,tcl检测反应,反应完毕,过滤,浓缩,烘干得化合物ⅳ,收率95.6%,hplc纯度99.87%。
[0057]
实施例3
[0058]
将化合物ii(10.3g,0.05mol)加入甲基叔丁基醚(50ml)中,控温5~10℃,搅拌 10min,滴加化合物iii(11.90g,0.1mol),滴加完毕,继续保温搅拌1h,tcl检测反应,反应完毕,过滤,浓缩,烘干得化合物ⅳ,收率95.8%,hplc纯度99.78%。
[0059]
实施例4
[0060]
将化合物ii(10.3g,0.05mol)加入乙醚(50ml)中,控温30~35℃,搅拌10min,滴加化合物iii(6.54g,0.055mol),滴加完毕,继续保温搅拌1h,tcl检测反应,反应完毕,过滤,浓缩,烘干得化合物ⅳ,收率92.2%,hplc纯度99.81%。
[0061]
实施例5
[0062]
将化合物ii(10.3g,0.05mol)加入四氢呋喃(50ml)中,控温35~40℃,搅拌10min,滴加化合物iii(12.50g,0.105mol),滴加完毕,继续保温搅拌1h,tcl检测反应,反应完毕,过滤,浓缩,烘干得化合物ⅳ,收率91.8%,hplc纯度99.66%。
[0063]
化合物vi的制备
[0064]
实施例6
[0065]
将化合物v(11.10g,0.05mol)加入到四氢呋喃(200ml)中,搅拌,分批缓慢加入氢化钠(2.4g,0.1mol),加入完毕,温度至-10℃,继续保温搅拌,反应完毕;将化合物iv (14.69g,0.065)滴加到反应体系,滴加完毕,控温-20℃搅拌;反应结束,缓慢加入2mol/l 稀硫酸调节ph=6~7;减压蒸除四氢呋喃,残余物加入二氯甲烷(100ml)和水(50ml)萃取,收集有机层,加入无水硫酸钠干燥,过滤,旋蒸,干燥得化合物vi,收率98.6%,hplc 纯度99.87%。
[0066]
实施例7
[0067]
将化合物v(11.10g,0.05mol)加入到乙醚(200ml)中,搅拌,分批缓慢加入氢化锂 (0.80g,0.1mol),加入完毕,温度至-10℃,继续保温搅拌,反应完毕;将化合物iv(13.56g, 0.06)滴加到反应体系,滴加完毕,控温-20℃搅拌;反应结束,缓慢加入2mol/l稀硫酸调节ph=5~6。;减压蒸除四氢呋喃,残余物加入二氯甲烷(100ml)和水(50ml)萃取,收集有机层,加入无水硫酸钠干燥,过滤,旋蒸,干燥得化合物vi,收率95.4%,hplc纯度99.78%。
[0068]
实施例8
[0069]
将化合物v(11.10g,0.05mol)加入到乙腈(200ml)中,搅拌,分批缓慢加入二异丙基氨基锂(10.71g,0.1mol),加入完毕,温度至-10℃,继续保温搅拌,反应完毕;将化合物iv(12.43g,0.06)滴加到反应体系,滴加完毕,控温-20℃搅拌;反应结束,缓慢加入 2mol/l稀硫酸调节ph=5~6;减压蒸除四氢呋喃,残余物加入三氯甲烷(100ml)和水(50ml) 萃取,收集有机层,加入无水硫酸钠干燥,过滤,旋蒸,干燥得化合物vi,收率92.6%, hplc纯度99.72%。
[0070]
实施例9
[0071]
将化合物v(11.10g,0.05mol)加入到甲基叔丁基醚(200ml)中,搅拌,分批缓慢加入双(三甲基硅基)氨基锂(16.73g,0.1mol),加入完毕,温度至-10℃,继续保温搅拌,反应完毕;将化合物iv(22.60g,0.1)滴加到反应体系,滴加完毕,控温-20℃搅拌;反应结束,缓慢加入2mol/l稀硫酸调节ph=6~7;减压蒸除四氢呋喃,残余物加入乙酸乙酯 (100ml)和水(50ml)萃取,收集有机层,加入无水硫酸钠干燥,过滤,旋蒸,干燥得化合物vi,收率96.6%,hplc纯度99.82%。
[0072]
实施例10
[0073]
将化合物v(11.10g,0.05mol)加入到乙醚(200ml)中,搅拌,分批缓慢加入双(三甲基硅基)氨基钠(18.37g,0.1mol),加入完毕,温度至-10℃,继续保温搅拌,反应完毕;将化合物iv(23.73g,0.105)滴加到反应体系,滴加完毕,控温-20℃搅拌;反应结束,缓慢加入2mol/l稀硫酸调节ph=5~6;减压蒸除四氢呋喃,残余物加入乙酸乙酯(100ml) 和水(50ml)萃取,收集有机层,加入无水硫酸钠干燥,过滤,旋蒸,干燥得化合物vi,收率91.2%,hplc纯度99.71%。
[0074]
实施例11
[0075]
将化合物v(11.10g,0.05mol)加入到四氢呋喃(200ml)中,搅拌,分批缓慢加入氢化钠(1.80g,0.075mol),加入完毕,温度至-20℃,继续保温搅拌,反应完毕;将化合物 iv(14.69g,0.065)滴加到反应体系,滴加完毕,控温-30℃搅拌;反应结束,缓慢加入 2mol/l稀硫酸调节ph=6~7;减压蒸除四氢呋喃,残余物加入二氯甲烷(100ml)和水(50ml) 萃取,收集有机层,加入无水硫酸钠干燥,过滤,旋蒸,干燥得化合物vi,收率96.4%, hplc纯度99.78%。
[0076]
实施例12
[0077]
将化合物v(11.10g,0.05mol)加入到四氢呋喃(200ml)中,搅拌,分批缓慢加入氢化钠(1.68g,0.07mol),加入完毕,温度至-25℃,继续保温搅拌,反应完毕;将化合物iv (14.69g,0.065)滴加到反应体系,滴加完毕,控温-35℃搅拌;反应结束,缓慢加入2mol/l 稀硫酸调节ph=6~7;减压蒸除四氢呋喃,残余物加入二氯甲烷(100ml)和水(50ml)萃取,收集有机层,加入无水硫酸钠干燥,过滤,旋蒸,干燥得化合物vi,收率93.5%,hplc 纯度
99.71%。
[0078]
实施例13
[0079]
将化合物v(11.10g,0.05mol)加入到四氢呋喃(200ml)中,搅拌,分批缓慢加入氢化钠(3.0g,0.125mol),加入完毕,温度至-5℃,继续保温搅拌,反应完毕;将化合物iv (14.69g,0.065)滴加到反应体系,滴加完毕,控温-10℃搅拌;反应结束,缓慢加入2mol/l 稀硫酸调节ph=6~7;减压蒸除四氢呋喃,残余物加入二氯甲烷(100ml)和水(50ml)萃取,收集有机层,加入无水硫酸钠干燥,过滤,旋蒸,干燥得化合物vi,收率96.6%,hplc 纯度99.79%。
[0080]
实施例14
[0081]
将化合物v(11.10g,0.05mol)加入到四氢呋喃(200ml)中,搅拌,分批缓慢加入氢化钠(3.12g,0.13mol),加入完毕,温度至0℃,继续保温搅拌,反应完毕;将化合物iv (14.69g,0.065)滴加到反应体系,滴加完毕,控温-5℃搅拌;反应结束,缓慢加入2mol/l 稀硫酸调节ph=6~7;减压蒸除四氢呋喃,残余物加入二氯甲烷(100ml)和水(50ml)萃取,收集有机层,加入无水硫酸钠干燥,过滤,旋蒸,干燥得化合物vi,收率90.6%,hplc 纯度99.69%。
[0082]
化合物i的制备
[0083]
实施例15
[0084]
将化合物vi(18.4g,0.0448mol)、10%氨的甲醇溶液(30.5g,0.179mol)和甲醇(110 ml)加入250ml高压釜,氨气保护,升温至60℃反应,tcl检测反应;反应结束,冷却至室温,减压蒸除溶剂,加入二氯甲烷(150ml)和水(50ml)萃取,收集有机层,加入无水硫酸钠干燥,过滤,旋蒸,干燥得化合物i,收率98.8%,hplc纯度99.92%。
[0085]
实施例16
[0086]
将化合物vi(18.4g,0.0448mol)、10%氨的甲醇溶液(15.23g,0.09mol)和四氢呋喃(110ml)加入250ml高压釜,氨气保护,升温至55℃反应,tcl检测反应;反应结束,冷却至室温,减压蒸除溶剂,加入三氯甲烷(150ml)和水(50ml)萃取,收集有机层,加入无水硫酸钠干燥,过滤,旋蒸,干燥得化合物i,收率95.8%,hplc纯度99.88%。
[0087]
实施例17
[0088]
将化合物vi(18.4g,0.0448mol)、10%氨的甲醇溶液(14.47g,0.085mol)和甲苯(110 ml)加入250ml高压釜,氨气保护,升温至50℃反应,tcl检测反应;反应结束,冷却至室温,减压蒸除溶剂,加入乙酸乙酯(150ml)和水(50ml)萃取,收集有机层,加入无水硫酸钠干燥,过滤,旋蒸,干燥得化合物i,收率92.3%,hplc纯度99.81%。
[0089]
实施例18
[0090]
将化合物vi(18.4g,0.0448mol)、10%氨的乙醇溶液(38.13g,0.224mol)和乙醇(110 ml)加入250ml高压釜,氨气保护,升温至75℃反应,tcl检测反应;反应结束,冷却至室温,减压蒸除溶剂,加入乙酸乙酯(150ml)和水(50ml)萃取,收集有机层,加入无水硫酸钠干燥,过滤,旋蒸,干燥得化合物i,收率96.6%,hplc纯度99.84%。
[0091]
实施例19
[0092]
将化合物vi(18.4g,0.0448mol)、10%氨的甲醇溶液39.15g,0.23mol)和甲醇(110 ml)加入250ml高压釜,氨气保护,升温至53℃反应,tcl检测反应;反应结束,冷却至室温,减
压蒸除溶剂,加入乙酸乙酯(150ml)和水(50ml)萃取,收集有机层,加入无水硫酸钠干燥,过滤,旋蒸,干燥得化合物i,收率91.8%,hplc纯度99.75%。
[0093]
实施例20
[0094]
将化合物vi(18.4g,0.0448mol)、10%氨的乙醇溶液(30.22g,0.179mol)和甲醇(110 ml)加入250ml高压釜,氨气保护,升温至65℃反应,tcl检测反应;反应结束,冷却至室温,减压蒸除溶剂,加入乙酸乙酯(150ml)和水(50ml)萃取,收集有机层,加入无水硫酸钠干燥,过滤,旋蒸,干燥得化合物i,收率90.2%,hplc纯度99.75%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1