一种具有抗炎作用的化合物及其制备方法和应用与流程
本发明涉及医药技术领域,尤其涉及一种具有抗炎作用的化合物及其制备方法和应用。
背景技术:
目前,临床上的解热药物主要包括非甾体类抗炎药和类固醇解热药。前者包括如水杨酸类、苯胺类、p比哩酮类以及其他抗炎有机酸类,这些药物共同的作用机制是通过抑制环氧化酶,减少前列腺素(prostaglandin,pg)合成,使体温调节中枢的sp恢复正常而产生解热作用,但是这些药物同时抑制了胃粘膜pg的合成,增加了胃酸分泌,削弱了屏障作用,导致胃肠道的不良反应,甚至引起胃粘膜损伤严重出现胃、十二指肠出血和馈病。传统中药由于其成分多,可能涉及多作用靶点发挥解热作用,且研究表明其降温作用稳定而持久。临床上,中药的解热抗炎药物大多为复方,由于其成分复杂,很难阐明其物质基础及作用机制,所以,从具有解热药效的单一药材入手,阐明其发挥作用的部位,进而寻找有效的成分,为开发成分明确、质量可控、安全有效的解热抗炎药物具有重要的意义,为临床用药提供更广泛的选择。
技术实现要素:
有鉴于此,本发明提供一种基于中药分离的一种具有抗炎作用的化合物及其制备方法和应用。
具体地,本发明提出了一种具有抗炎作用的化合物或者其药学上可接受的盐、溶剂化物、异构体、前药分子或代谢物,该化合物的化学式为6-羟基-4-甲氧基-5,7-二甲基橙酮。
具体地,所述药学上可接受的盐包括盐酸盐、硫酸盐、枸橼酸盐、苯磺酸盐、氢溴酸盐、氢氟酸盐、磷酸盐、乙酸盐、丙酸盐、丁二酸盐、草酸盐、苹果酸盐、琥珀酸盐、富马酸盐、马来酸盐、酒石酸盐或三氟乙酸盐。
进一步地,所述异构体选自(e)-6-羟基-4-甲氧基-5,7-二甲基橙酮。其中,该异构体可以采用水翁花提取分离的方式获得,也可以采用(z)-6-羟基-4-甲氧基-5,7-二甲基橙酮经光照转化后制备液相分离的方式获得。
具体地,所述采用(z)-6-羟基-4-甲氧基-5,7-二甲基橙酮经光照转化后制备液相分离的方式包括如下步骤:
步骤1:取(z)-6-羟基-4-甲氧基-5,7-二甲基橙酮用溶剂溶解,将溶液放置光照下,光照后的溶液减压回收溶剂,得浸膏;
步骤2:将步骤1中浸膏用乙酸乙酯溶解,经制备液相分离,得到式i所示化合物。
进一步地,上述过程中,转化溶剂可选自甲醇、乙醇、甲醇-水、乙醇-水,优选为乙醇;所述光照强度为100~4500lux,优选1500lux;制备液相分离中所用流动相为乙酸乙酯-正己烷溶液或乙酸乙酯-石油醚,优选流动相为乙酸乙酯-正己烷,体积比为1:(4~8),优选为1:5。
本发明还提出了如前任一所述的化合物或者其药学上可接受的盐、溶剂化物、异构体、前药分子或代谢物在制备抗炎或镇痛药物中的应用。
本发明还提出了包括如前任一所述的化合物或者其药学上可接受的盐、溶剂化物、异构体、前药分子或代谢物的药物。
具体地,前述药物包括药学上可接受的辅料。其中,该辅料可根据剂型和实际情况进行恰当选择,例如常用的辅料有淀粉、淀粉浆、蔗糖、糊精、低取代羟丙基纤维素、微粉硅胶、羧甲基淀粉钠、滑石粉、硬脂酸镁、聚山梨酯、聚乙二醇、注射用大豆磷脂和注射用甘油等;利用本发明得到化合物,制备所需药物的各种剂型时,可以按照药剂学领域的常规生产方法制备。如将该化合物或者其药学上可接受的盐、溶剂化物、异构体、前药分子或代谢物与一种或多种载体混合,然后制成相应的剂型。
进一步地,所述药物为口服给药剂型、注射给药剂型或外用给药制剂。
具体地,所述药物包括片剂、胶囊剂、软胶囊、凝胶剂、口服剂、混悬剂、冲剂、贴剂、软膏、合剂、丸剂、散剂、注射剂、输液剂、冻干注射剂、静脉乳剂、脂质体注射剂、栓剂、缓释制剂或控释制剂。
本发明还提出了化合物6-羟基-4-甲氧基-5,7-二甲基橙酮的制备方法,该方法包括:
步骤1:获得水翁花的提取液;其中,本发明对水翁花提取的方法没有特殊限制,本领域公知的用于溶剂提取中草药成分的方法均可;
步骤2:将步骤1制得的提取液调节乙醇体积浓度至50%~70%,静置,取上清液经大孔树脂吸附并用50%~70%浓度乙醇淋洗;然后用80%~95%乙醇洗脱,收集洗脱液减压浓缩,得到浸膏;
步骤3:将步骤2制得的浸膏用乙酸乙酯溶解,加入石油醚搅拌;取上清液回收溶剂,得到浸膏;
步骤4:将步骤3制得的浸膏,经制备型液相色谱分离。
进一步地,所述步骤1中,优选用乙醇提取得到水翁花的乙醇提取液,所述乙醇优选为60%~90%乙醇水溶液,更优选为70%~85%乙醇水溶液;所述水翁花与乙醇水溶液的重量比为1:(7~10);所述提取的次数为1~3次,优选为2次;所述提取的时间优选为1~3小时,更优选为1.5~2.0小时。
进一步地,所述步骤2中,所述大孔树脂为弱极性或非极性树脂,所述弱极性树脂为ab-8、dm130,所述非极性树脂为d101、hpd100、hp20,优选d101型大孔树脂。
进一步地,所述步骤3中,乙酸乙酯与石油醚的用量体积比为1:(5~15),更优选为1:(8~12)。
进一步地,所述步骤4中,制备型液相色谱分离的流动相选自乙腈-0.1%甲酸水溶液。更具体地,制备型液相色谱分离的条件包括:流动相体积浓度为(40~60)%乙腈-0.1%甲酸水溶液,优选体积浓度为(45~55)%乙腈-0.1%甲酸水溶液;流速为5~60ml/min,优选为15~30ml/min;检测波长为360nm。
进一步地,化合物6-羟基-4-甲氧基-5,7-二甲基橙酮的制备方法包括如下步骤:
步骤1:水翁花加入10倍量的体积浓度为70%乙醇水溶液,提取2次,每次2h,得提取液;
步骤2:将步骤1制得的提取液加入纯化水调节乙醇体积浓度至60%,静置过夜,取上清液经d101大孔树脂吸附,然后用60%乙醇洗脱4个柱体积除去杂质,然后用90%乙醇洗脱4个柱体积,收集90%乙醇洗脱液减压浓缩回收溶剂,得到浸膏;
步骤3:将步骤2制得的浸膏用乙酸乙酯溶解,加入石油醚搅拌,其中乙酸乙酯与石油醚用量的体积比为1:10;取上清液回收溶剂,得到浸膏;
步骤4:将步骤3制得的浸膏经制备型hplc分离,以体积浓度为52%乙腈-0.1%甲酸水溶液为流动相,检测波长为360nm,流速17ml/min,分离所得溶液干燥。
本发明通过对水翁花进行提取,得到浸膏;然后将得到的浸膏进行分离,选择特定出峰时间的化合物,得到一种橙酮化合物;或通过(z)-6-羟基-4-甲氧基-5,7-二甲基橙酮在光照下进行转化,也可得到该橙酮化合物;通过体外活性筛选发现该化合物对脂多糖(lps)诱导的小鼠raw264.7巨噬细胞pge2的产生具有较强的抑制作用,活性优于布洛芬,该化合物具有一定的抗炎活性;通过小鼠醋酸扭体试验,发现给化合物具有一定的镇痛作用。
附图说明
图1为本发明实施例1制得的化合物的esi-ms谱图;
图2为本发明实施例1制得的化合物的1h-nmr谱;
图3为本发明实施例1制得的化合物的13c-nmr谱;
图4为本发明实施例1制得的化合物的hsqc谱;
图5为本发明实施例1制得的化合物的hmbc谱;
图6为本发明实施例1制得的化合物的主要hmbc相关。
具体实施方式
下面将结合本发明实施例的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明实施例的水翁花始载于《岭南采药录》,为桃金娘科水翁属植物水翁cleistocalyxrculatus(roxb.)merr.etperry的干燥幼嫩花蕾,又名水雍花、大蛇药、水翁仔、水榕花、水翁、酒翁、水榕树、水香;性寒,味苦,归脾、胃经;能清热解毒,去湿消滞;主用于夏天暑湿食滞所致的发热,咽干,口渴腹胀或呕吐泄泻。水翁花在民间应用广泛,属于广东地产药材,夏季常作凉茶以解暑,是“清热凉茶”等中药的主要原料之一。
橙酮是植物体内的次生代谢物产物,属于类黄酮,在自然界分布比较少,含量都很低,大多存在于玄参科、菊科、苦苣苔科以及单子叶植物中的莎草科。
实施例1本发明化合物的制备
1)干燥的水翁花5kg,加入10倍量的体积浓度为70%乙醇水溶液,提取2次,每次2h,得提取液;
2)取步骤1)的提取液加入适量的纯化水调节乙醇体积浓度至60%,静置过夜,取上清液经d101大孔树脂吸附,然后用60%乙醇洗脱4个柱体积除去杂质,然后用90%乙醇洗脱4个柱体积,收集90%乙醇洗脱液减压浓缩回收溶剂,得到浸膏;
3)取步骤2)中的浸膏用乙酸乙酯溶解,加入石油醚搅拌,其中乙酸乙酯与石油醚用量的体积比为1:10;取上清液回收溶剂,得到浸膏;
4)取步骤3)中的浸膏经制备型hplc分离,以体积浓度为52%乙腈-0.1%甲酸水溶液为流动相,检测波长为360nm,流速17ml/min,分离所得溶液干燥,得到本发明所述化合物28mg,纯度为98.9%。
实施例1得到的化合物为黄色无定型粉末。
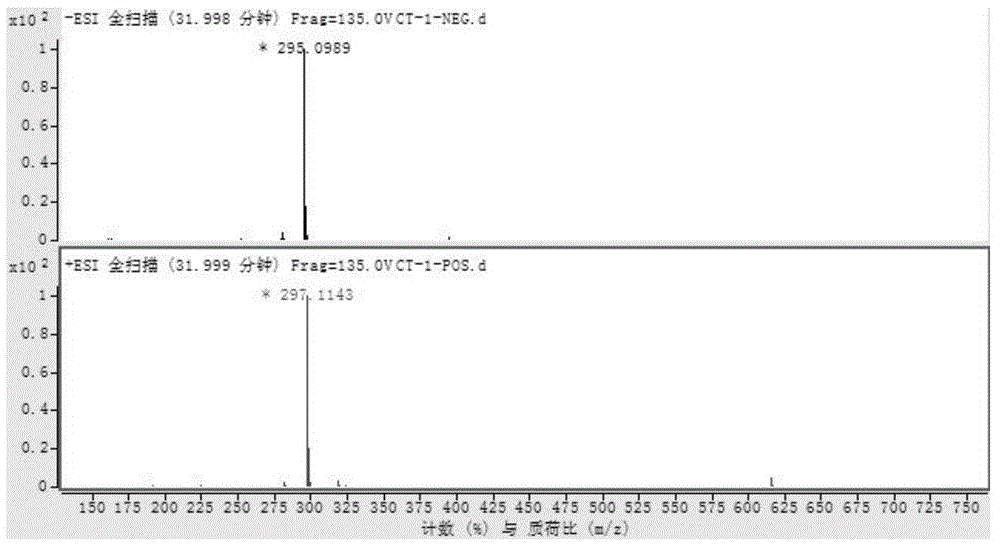
对上述化合物进行结构分析,高分辨质谱hr-esi-ms给出m/z295.0989[m-h]-(计算值为295.0976),m/z297.1143[m+h]+(计算值为297.1121),提示化合物分子量为296,结合元素分析及13c-nmr谱和1h-nmr谱推断该化合物分子式为c18h16o4。
对实施例1得到的化合物进行结构鉴定,结果见图1~6,图1为本发明实施例1制得的化合物的esi-ms谱图,图2为本发明实施例1制得的化合物的1h-nmr谱图,图3为本发明实施例1制得的化合物的13c-nmr谱图,图4为本发明实施例1制得的化合物的hsqc谱图,图5为本发明实施例1制得的化合物的hmbc谱图,图6为本发明实施例1制得的化合物的主要hmbc相关。
通过对图1~图6进行分析,本化合物的1h-nmr(dmso-d6,400mhz)谱(见图2),给出1组典型的单取代苯环质子信号δ8.20-8.12(2h,m,h-2′,6′),δ7.41-7.35(2h,m,h-3′,5′),δ7.45-7.41(1h,m,h-4′);1个烯质子信号δ6.98(1h,s,h-10);2个甲基单峰质子信号δ2.08(3h,s,h-12)和δ1.99(3h,s,h-11);和1个甲氧基质子信号δh3.93(3h,s)。
13c-nmr(dmso-d6,400mhz)谱中(见图3),共给出18个碳信号,结合1h-nmr谱数据推测该化合物为橙酮类化合物;其中橙酮母核碳信号15个,δ177.42为橙酮母核3位的特征羰基碳信号;δ118.21为烯碳(=ch—)信号;δ8.78和δ7.89为两个甲基碳信号,δ60.96为甲氧基碳信号。
结合hsqc图谱(见图4)以及hmbc图谱(见图5)对具有相关关系的碳信号与质子信号进行归属,甲基质子δ1.99与c-4、c-5和c-6相关,推测该甲基连接在c-5上;甲基质子δ2.08与c-6、c-7、c-8相关,推测该甲基与c-7相连;甲氧基质子δ3.93与c-4相关,推测甲氧基与c-4相连。
由此确定,本发明得到的化合物化学式为6-羟基-4-甲氧基-5,7-二甲基橙酮。此外,进一步对橙酮类化合物双键的构型进行分析,一般认为双键(=ch—)的碳原子化学位移值在δ110,质子在δ6.7左右时,为z构型;碳原子化学位移在δ120左右,质子化学位移δ7.0左右时,双键为e构型;本化合物双键(=ch—)的碳原子δ118.21,质子δ6.98,判断双键为e构型。
本发明化合物(e)-6-hydroxy-4-methoxy-5,7-dimethylaurone,中文名为(e)-6-羟基-4-甲氧基-5,7-二甲基橙酮。其所有的碳氢信号归属见表1,表1为式i所示化合物的各个碳和氢的归属。
表1化合物的核磁数据(dmso-d6,1h-nmr400mhz,13c-nmr400mhz)
实施例2本发明化合物的制备
1)干燥的水翁花5kg,加入7倍量的体积浓度为80%乙醇水溶液,提取3次,每次1.5h,得提取液;
2)取步骤1)的提取液加入适量的纯化水调节乙醇体积浓度至50%,静置过夜,取上清液经hp-20大孔树脂吸附,然后用70%乙醇洗脱4个柱体积除去杂质,然后用95%乙醇洗脱4个柱体积,收集95%乙醇洗脱液减压浓缩回收溶剂,得到浸膏;
3)取步骤2)中的浸膏用乙酸乙酯溶解,加入石油醚搅拌,其中乙酸乙酯与石油醚用量的体积比为1:15;取上清液回收溶剂,得到浸膏;
4)取步骤3)中的浸膏经制备型hplc分离,以体积浓度为40%乙腈-0.1%甲酸水溶液为流动相,检测波长为360nm,流速60ml/min,分离所得溶液干燥,得到本发明所述的化合物29mg,纯度为98.5%。
按照实施例1提供的理化和结构分析方法对化合物进行分析可知,实施例2得到的化合物的结构为式ⅰ所示的化合物。
实施例3本发明化合物的制备
1)干燥的水翁花5kg,加入8倍量的体积浓度为90%乙醇水溶液,提取1次,每次1h,得提取液;
2)取步骤1)的提取液静置过夜,取上清液经ab-8大孔树脂吸附,然后用50%乙醇洗脱4个柱体积除去杂质,然后用80%乙醇洗脱4个柱体积,收集80%乙醇洗脱液减压浓缩回收溶剂,得到浸膏;
3)取步骤2)中的浸膏用乙酸乙酯溶解,加入石油醚搅拌,其中乙酸乙酯与石油醚用量的体积比为1:5;取上清液回收溶剂,得到浸膏;
4)取步骤3)中的浸膏经制备型hplc分离,以体积浓度为60%乙腈-0.1%甲酸水溶液为流动相,检测波长为360nm,流速5ml/min,分离所得溶液干燥,得到本发明所述的化合物30mg,纯度为99.1%。
按照实施例1提供的理化和结构分析方法对化合物进行分析可知,实施例3得到的化合物的结构为式ⅰ所示的化合物。
实施例4本发明化合物的制备
1):取500mg(z)-6-羟基-4-甲氧基-5,7-二甲基橙酮用500ml乙醇溶解,得溶液;将溶液放置光照强度为1500lux的光照下,光照4小时后将溶液减压回收溶剂,得浸膏;
2)取步骤1)中的浸膏用乙酸乙酯溶解,经grace制备液相分离,以乙酸乙酯-正己烷体积比为1:5为流动相,检测波长为360nm,流速15ml/min,分离所得溶液干燥,得到本发明所述的化合物206mg,纯度为98.4%。
按照实施例1提供的理化和结构分析方法对化合物进行分析可知,实施例4得到的化合物的结构为式ⅰ所示的化合物。
实施例5本发明化合物的制备
1):取500mg(z)-6-羟基-4-甲氧基-5,7-二甲基橙酮用500ml甲醇溶解,得溶液;将溶液放置光照强度为100lux的光照下,光照8小时后将溶液减压回收溶剂,得浸膏;
2)取步骤1)中的浸膏用乙酸乙酯溶解,经grace制备液相分离,以乙酸乙酯-正己烷体积比为1:4为流动相,检测波长为360nm,流速10ml/min,分离所得溶液干燥,得到本发明所述的化合物194mg,纯度为98.7%。
按照实施例1提供的理化和结构分析方法对化合物进行分析可知,实施例5得到的化合物的结构为式ⅰ所示的化合物。
实施例6:本发明化合物的制备
1):取500mg(z)-6-羟基-4-甲氧基-5,7-二甲基橙酮用500ml95%乙醇溶解,得溶液;将溶液放置光照强度为4500lux的光照下,光照2小时后将溶液减压回收溶剂,得浸膏;
2)取步骤1)中的浸膏用乙酸乙酯溶解,经grace制备液相分离,以乙酸乙酯-正己烷体积比为1:8为流动相,检测波长为360nm,流速20ml/min,分离所得溶液干燥,得到本发明所述的化合物208mg,纯度为98.6%。
按照实施例1提供的理化和结构分析方法对化合物进行分析可知,实施例6得到的化合物的结构为式ⅰ所示的化合物。
实施例7本发明化合物的抗炎活性
1.材料
1.1药物实施例1所得式i所示化合物;
1.2阳性对照药布洛芬(中国食品药品检定研究院,批号:201707);
1.3试剂细胞株:小鼠巨噬细胞系raw264.7,来源于中医科学院,江苏康缘药业股份有限公司培养;脂多糖(lps):南京大治生物科技有限公司;前列腺素e2(pge2)elisa试剂盒:恩佐生命科学,批号:01071907c。
2.实验方法与步骤
2.1细胞培养:raw264.7细胞接种于细胞培养瓶中,加入含10%灭活胎牛血清的dmem培养基,于37℃,5%co2的培养箱中培养,隔天更换培养基1次,取对数生长期的细胞进行后续实验。
2.2细胞活力检测:通过mtt法检测细胞活力,实验分为空白组、溶剂对照组、布洛芬组、式i所示化合物组;实验重复3次。
2.3elisa法检测pge2的含量:调整raw264.7细胞密度为1×105个/ml,接种于24孔细胞培养板中,每孔500μl于37℃,5%co2的培养箱中培养。24h后弃去上清,实验分为空白组、溶剂对照组(加入含0.1%dmso的dmem培养基)、模型组、给药组(加入式i所示化合物、布洛芬的dmso溶液)于37℃,5%co2的培养箱中培养1h后,除空白组外,每孔加入lps(终浓度1μg/ml)刺激24h后,收集上清。按照elisa试剂盒说明书检测pge2的含量,并按照下列公式计算抑制率,采用graphpadprismtm5.0软件进行统计学分析,比较ic50值。
3.实验结果
细胞活力检测结果显示,在0.1-10000nmol/l范围内式i所示化合物对raw264.7细胞的生长无毒性作用。
抗炎活性测试结果表明,式i所示化合物对pge2抑制的ic50分别为8.8nm,阳性对照布洛芬对pge2抑制的ic50为68.7nm;与布洛芬相比,式i所示化合物有更好的抗炎活性。
4.结论
本发明化合物对脂多糖(lps)诱导的小鼠raw264.7巨噬细胞pge2的产生具有较强的抑制作用,活性优于布洛芬,该化合物具有一定的抗炎活性。
实施例8本发明化合物的镇痛作用
1.材料
1.1药物实施例1所得式i所示化合物
1.2阳性对照药
阿司匹林(中国食品药品检定研究院,批号:201706)
1.3试验动物
小鼠,60只、雌雄各半,体重19~21g。
2.实验方法与步骤
2.1阳性药、式i所示化合物及阴性制剂的配制
阿司匹林溶液的配制:精密称取阿司匹林粉末1.35g,置于西林瓶中,加100ml0.5%cmc-na溶液超声溶解,即得浓度为13.5mg/ml的阿司匹林溶液;同法配制阴性制剂1,以上制剂2-4℃保存待用。
式i所示化合物溶液的配制:精密称取式i所示化合物15mg、羟丙基倍他环糊精150mg、碳酸钠10.5mg,用15ml75%乙醇加热溶解,浓缩回收溶剂至干膏,加入10ml生理盐水溶解,过0.22微米双层滤膜(水膜);即得浓度为1.5mg/ml的式i所示化合物溶液;同法配制阴性制剂2,以上制剂2-4℃保存待用。
2.2造模及给药
小鼠,雌雄各半,体重19~21g,随机分为正常对照组、阿司匹林组(270mg/kg)、式i所示化合物高剂量组(15mg/kg)、式i所示化合物低剂量组(7.5mg/kg)及阴性制剂组1、阴性制剂组2,每组12只。正常对照组、阿司匹林组(0.27g·kg-1)阴性制剂组1采用灌胃给药,式i所示化合物高、低剂量组及阴性制剂组2采用腹腔注射给药。在干预60min后,腹腔注射0.6%冰醋酸10ml·kg-1,观察记录30min内小鼠出现扭体反应鼠数和扭体次数及出现扭体的反应时间。
抑制率=(对照组扭体数-给药组扭体数)/对照组扭体数×100%
2.3数据处理
所有数据以
3实验结果
表2橙酮对醋酸致小鼠扭体反应的抑制作用(
与正常对照组相比,*p<0.05,**p<0.01.
如表2所示,与正常对照组比较,阿司匹林组表现出一定的镇痛作用(p<0.01),式i所示化合物阴性制剂组未表现出镇痛作用(p>0.05),但式i所示化合物高剂量组(p<0.01)及低剂量组(p<0.05)表现出一定的镇痛作用。结论:式i所示化合物具有一定的外周镇痛作用。
实施例9式i所示结构的化合物制备片剂药物
将200g式i所示化合物和60g淀粉、7.5g羧甲基淀粉钠、0.8g滑石粉、50g糊精、0.8g硬脂酸镁及适量10%淀粉浆适混合,按照常规方法制成新化合物片剂1000片。每日3次,每次1片。
实施例10式i所示结构的化合物制备胶囊剂药物
将200g式i所示化合物和50g淀粉、6.5g微粉硅胶、6g低取代羟丙基纤维素、1.5g硬脂酸镁及适量10%淀粉浆混合,装入胶囊,得到新化合物的胶囊制剂1000粒。每日3次,每次1粒。
实施例11:式i所示结构的化合物制备颗粒剂药物
将200g式i所示化合物和600g糊精及800g蔗糖混合,按照常规方法制成1000包新化合物颗粒剂。每日3次,每次1粒。
实施例12:式i所示结构的化合物制备丸剂药物
将200g式i所示化合物和20g聚乙二醇-6000、80g聚山梨酯-80、适量液状石蜡混合,按照常规方法制成新化合物丸剂1000粒。每日3次,每次1粒。
实施例13:式i所示结构的化合物制备注射剂药物
将100g式i所示化合物和20g注射用大豆磷脂、30g注射用甘油,注射用水定容至1000ml,按照常规方法制成新化合物注射剂1000支。每日1次,每次1支,至少采用250ml5%葡萄糖注射液稀释后静脉滴注。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!