一种弓形虫wx2基因缺失株、构建方法及用途与流程
本发明涉及一种弓形虫基因缺失株,具体涉及一种弓形虫wx2基因缺失株、构建方法及用途。属于基因工程
技术领域:
。
背景技术:
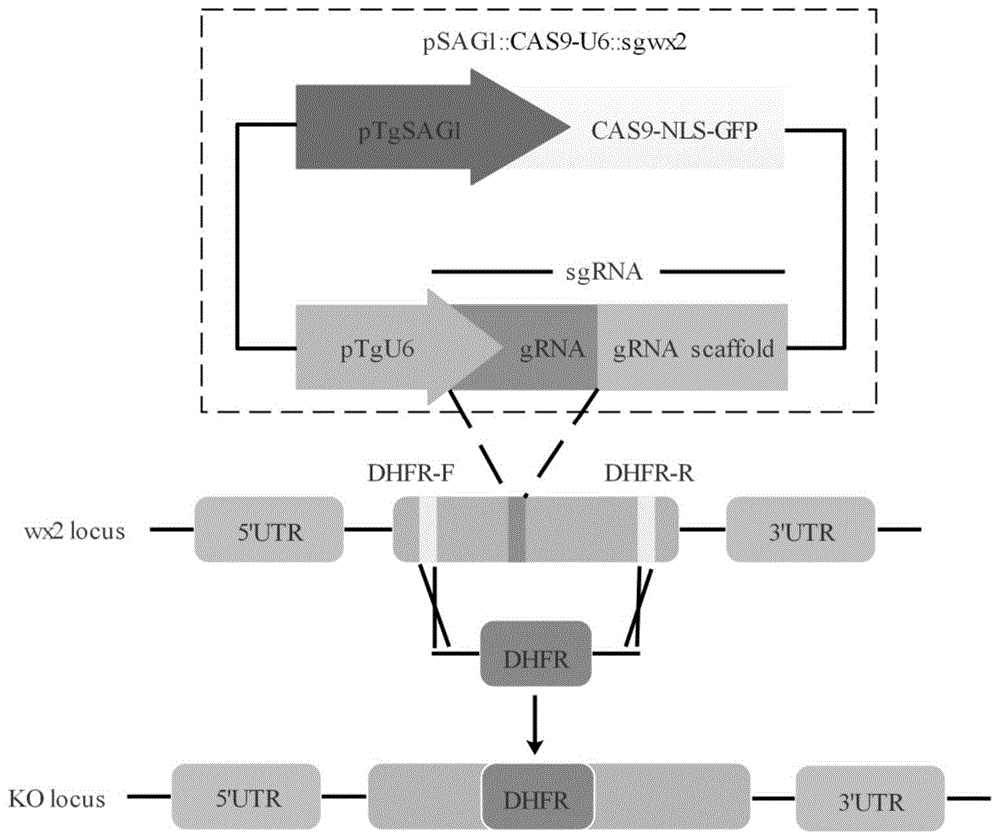
:弓形虫(toxoplasma)在生物界的分类隶属于顶复门、孢子虫纲、真球虫目、肉孢子虫科、弓形虫属。弓形虫寄生广泛,几乎可以感染所有有核细胞。弓形虫需要中间宿主和终末宿主的参与,来完成整个生活史。弓形虫是一种机会致病性原虫,传播途径主要通过粪口、生食或半生食肉类、垂直传播,全球有三分之一以上的人感染该病原体。弓形虫不仅危害人类的身体健康,也不利于动物的繁殖和畜牧业的发展。弓形虫生活史复杂,宿主十分广泛,危害严重,但目前还没有很好的药物治疗,尤其是针对慢性期包囊的治疗。人感染弓形虫病后,通常使用磺胺嘧啶类治疗,这类药物副作用较大,对胎儿也有致畸作用。此外,目前还没有药物能够杀死包囊中的缓殖子。虽然一种商品化减毒疫苗(intervetb.v.)已在兽医行业小范围使用,以避免羊的先天性感染。但其可能的副作用和高昂的成本限制了这一商品化疫苗的推广;同时人用弓形虫疫苗的研究仍然没有成功。因此,目前还没有有效的措施来防治弓形虫病。申请人的前期研究已初步证明wx2为弓形虫功能膜分子,参与机体的细胞免疫应答,并且与弓形虫的毒力相关。但该分子的大部分功能及机制尚不明确。因此,需要进一步深入研究wx2基因的功能,为研发弓形虫药物的作用靶点和弓形虫疫苗提供参考。技术实现要素:本发明的目的是为克服上述现有技术的不足,提供一种弓形虫wx2基因缺失株、构建方法及用途。为实现上述目的,本发明采用下述技术方案:1、一种弓形虫wx2基因缺失株,是缺失wx2基因的弓形虫rh株,wx2基因的核苷酸序列如seqidno.11所示。wx2基因:ggcacgagcgagcgtgttgcgaaggcaaaccgataaacagttcttgctctctgcctgttggaaaccatgttcttacgatggctgggctgttttcgcgcatcctcgtttgacgtaggttagtgacatgcttctactccgtgcttcgtcagtgaaaactttgtgttgccgaaacaagcaatgtgcctctactgggcaacttcctcttaacatgttcaaggtcgaaaaaaaaaaaaaaaaaaactcgagagatcttttgagcacaagaaggtacacgtttctttcccttctgctcgcctcggcggtaaggctggtgcgcatttgttgcacgcgcatctttccacatgaaaaaagagcgcgtatcgggataacgggagacagagcgaaaataaaagagggcgggtttcatttggaacaacgccaagacattgtttctgcaacatacaccgcgagaaactgctgcaccccgcagtagagattgaacttcaccggaatcgagtgtaatccacacgctagaccaggcctaaagacgtttacccagttcctaaccgggcgccgcgcttgtccttcgagaaaaaacttcatggaagaaaaaagctcacactttgtaccgacaaacggcaaacattagcgccatacgcaaaaggaaccgcatgcacacagctcggtaagctcctcctctcgacggtacctgacctaattcggtgcaccgagcgatagacaattgcggagacacatgtatatctgtatagaaggtacgcaaaaaggtaggatcacagacgcgcatgcaaaaacacattcctcacagacgcgatggcgtcctgcacctgcagtgccgcttcttcattcaccgattcacatgacattttccttcgaccggagagacgaaaacatccgtaaagcggtgagtgtcgttctacgtatcgcctctcgtcgccgaagagaagaaattcctccatactcaggactcttcggatttcctcagtttgcttctgcattccgtgttcccttcttcgtcgtctctcctccctcctccccatttctttgcttcctctctctcctttctaaccgtctctgcggctctccgccgtcgctccgttcctcgactttgcttctcggagtcgagcgcgtcgacccttcctcccggtttctctcttctctggatacgagcgaacgcgagctgaccgcgtcgcaccaccctcaaaacactcgcacggggtcccatgtgaggtacacacacgaagtatttttcctccggctgcacttgaacgtacacagaagtcaggcgacaccgtgagtgcagcaggcccgcggcttgggggagctccggcctctctaaacgaatctgaaaatgcgcgaaagaaaagcgacttgcccttcccgcacagtccgtggtgtggcgcgagcgacattccacgcctgggatttgcgtcatcacaaaactatcgaaccagcaaatgcacttgtttttcgccaccgcagagacacgtgagtcattgttgctctcgcttctttgcc,如seqidno.11所示。优选的,所述的一种弓形虫wx2基因缺失株具乙胺嘧啶药物抗性。2、上述一种弓形虫wx2基因缺失株的构建方法,具体步骤如下:(1)先以psag1::cas9-u6::sguprt载体为模板,核苷酸序列分别如seqidno.1和seqidno.2所示的上游引物sgwx2-f和下游引物sgwx2-r为引物进行pcr扩增,连接,得到psag1::cas9-u6::sgwx2质粒,即为crispr/cas敲除质粒;(2)再以puprt::dhfr-d载体为模板,核苷酸序列分别如seqidno.3和seqidno.4所示的上游引物wx2-dhfr-f和下游引物wx2-dhfr-r为引物进行pcr扩增,即得wx2-dhfr片段;(3)最后将步骤(1)所得crispr/cas敲除质粒、步骤(2)所得wx2-dhfr片段与弓形虫rh株制成的弓形虫悬液混合,电转,培养,筛选,获得所述的一种弓形虫wx2基因缺失株。优选的,步骤(1)的具体方法是:先利用psag1::cas9-u6::sguprt载体、核苷酸序列分别如seqidno.1和seqidno.2所示的上游引物sgwx2-f和下游引物sgwx2-r构建pcr反应体系,进行pcr扩增,回收pcr产物,并进行sgwx2粗产物消化;接着将步骤(1)所得消化产物进行连接,使得sgwx2由线性变为环状,得到psag1::cas9-u6::sgwx2质粒,即为crispr/cas敲除质粒。进一步优选的,所述pcr反应体系包含:q5热启动超保真2×mastermix12.5μl,10μm上游引物sgwx2-f1μl,10μm下游引物sgwx2-r1μl,psag1::cas9-u6::sguprt载体2μl,双蒸水8.5μl。进一步优选的,pcr扩增条件为:98℃预变性1min;98℃变性10s,55℃复性30s,72℃延伸5min,共25个循环;72℃再延伸2min。优选的,步骤(2)中,所述pcr反应体系包含:q5热启动超保真2×mastermix25μl,20μm上游引物wx2-dhfr-f1μl,20μm下游引物wx2-dhfr-r1μl,puprt::dhfr-d3μl,双蒸水20μl。优选的,步骤(2)中,pcr扩增条件为:98℃预变性3min;98℃变性10s,55℃复性30s,72℃延伸2min30s,共30个循环;72℃再延伸2min。优选的,步骤(3)的具体方法如下:(3-1)先吸取28μlcytomixbuffer于无菌离心管中,将20μgcrispr/cas9敲除质粒与2μgdhfr片段加入该离心管,使终体积为50μl,混匀,再加入250μl弓形虫悬液,用移液枪轻轻吹打混匀;(3-2)然后迅速转移到电转杯中,设置1500v5ms电击2次,将电击后的速殖子悬液转移至含新鲜2%fbsdmem培养基的hff细胞培养瓶中,在37℃5%co2培养箱中培养24小时;(3-3)最后弃去残留在细胞外的弓形虫速殖子,更换fbs浓度为2%dmem培养基继续培养,培养基中加入浓度为3μm的乙胺嘧啶,筛选,获得所述的一种弓形虫wx2基因缺失株。进一步优选的,筛选的具体方法是:先利用3μm的乙胺嘧啶筛选3~5代后,收集逸出的弓形虫速殖子,用血球计数板计数并调整浓度至3~5个/100μl;然后将速殖子接种至96孔板中,同时进行倍比稀释,培养单克隆;最后在温箱中培养一周后,镜下观察是否有单一虫斑,将单一的虫斑接种到24孔板中继续培养,待虫斑长满后,部分冻存在-80℃超低温冰箱用于提取dna。优选的,步骤(3)中,所述弓形虫悬液的制备方法如下:(a)先将冻存的弓形虫rh株接种至hff细胞,当有75%虫体逸出时,将细胞刮起并悬浮于培养基中,得细胞悬液;(b)然后对细胞悬液进行反复破碎,使虫体从细胞内充分逸出,去除细胞碎片,得虫体悬液;(c)再用cytomixbuffer对虫体悬液进行重悬,调整虫体浓度为2×107个/ml,即得所述的弓形虫悬液。3、上述一种弓形虫wx2基因缺失株在制备抗弓形虫疫苗中的用途。优选的,所述疫苗为减毒活疫苗。4、上述一种弓形虫wx2基因缺失株在抗弓形虫药物靶标分子筛选中的用途。5、一种抗弓形虫疫苗,其有效成分为上述的一种弓形虫wx2基因缺失株。本发明的有益效果:本发明通过构建弓形虫wx2基因缺失株,旨在揭示wx2基因在弓形虫中的生物学功能,为进一步作为药物靶标分子或减毒活疫苗的研究提供理论依据。本发明运用crispr/cas9技术,将弓形虫rh株的wx2基因进行敲除,成功获得具有乙胺嘧啶药物抗性的wx2基因缺失株。具体方法是通过构建crispr质粒psag1-cas9-u6-sgwx2和药物抗性片段wx2-dhfr,利用crispr质粒的靶向剪切和药物抗性片段的同源重组,经药物筛选单克隆和单克隆缺失株的鉴定,成功获得弓形虫rh株的wx2基因缺失株。本发明利用psag1::cas9-u6::sguprt质粒和puprt::dhfr-d质粒,首先构建了wx2基因特异的基因敲除质粒psag1-cas9-u6-sgwx2和含有特异同源臂的乙胺嘧啶抗性片段wx2-dhfr。基因沉默的部分位于wx2的外显子区域,确保wx2的功能完全缺失。sgwx2的碱基数目以20bp,位于外显子的中间区域。wx2基因被剪切后,利用同源重组的原理,wx2-dhfr会整合到目的基因并替换原有的片段,因此虫株在wx2基因缺失的同时,还会产生乙胺嘧啶抗性。由于整合的效率不是100%,因此可通过在培养基中添加乙胺嘧啶,通过有限稀释法筛选单克隆wx2基因缺失虫株。为确保所筛选单克隆为wx2缺失株,提取了mrna和dna,证实缺失株的wx2基因已成功敲除。采用噬斑实验得出,wx2基因缺失株和野生株的噬斑面积分别为10.3±4.9mm2和31.7±7.3mm2,表明wx2基因有利于弓形虫rh株的生长;入侵实验观察到,wx2基因缺失株和野生株的细胞入侵率分别为28%±3%和79%±8%,说明wx2基因参与弓形虫rh株入侵宿主细胞;胞内繁殖实验可以看出,与野生株相比,wx2基因缺失株在宿主细胞以及纳虫泡内的数量减少,说明wx2基因有利于弓形虫rh株的繁殖;进行小鼠体内攻击实验,记录被感染小鼠的存活时间,wx2基因缺失株组和野生株组攻击的小鼠存活时间相比,敲除株显著延长了攻击小鼠的生存时间,说明wx2基因与弓形虫rh株的毒力相关。本发明为弓形虫生长的分子机制的研究夯实基础,为弓形虫疫苗的研制和弓形虫药物靶标分子的研究提供了理论基础。附图说明图1为本发明的一种弓形虫wx2基因缺失株的构建过程示意图;图2为psag1::cas9-u6::sguprt质粒sgrna序列的替换琼脂糖凝胶电泳图,其中m为分子量标准,wx2为wx2基因片段替换后的敲除质粒pcr产物;图3为psag1::cas9-u6::sgwx2质粒琼脂糖凝胶电泳图,其中,m为分子量标准,2为完整的psag1::cas9-u6::sgwx2质粒,1、3、4为不完整的psag1::cas9-u6::sgwx2质粒;图4为psag1::cas9-u6::sgwx2测序结果比对;图5为dnfr片段琼脂糖凝胶电泳图;图6为线粒体基因rpl7的扩增琼脂糖凝胶电泳图;图7为wx2基因的扩增琼脂糖凝胶电泳图;图8为线粒体基因rpl7的扩增琼脂糖凝胶电泳图,其中,+表示rh株速殖子cdna,-为蒸馏水;图9为内含子基因片段的扩增琼脂糖凝胶电泳图;图10为wx2基因的扩增琼脂糖凝胶电泳图,其中,+表示rh株速殖子cdna,-为蒸馏水;图11为wx2缺失株(a)与野生株rh(b)所形成的噬斑;图12为wx2缺失株与野生株rh所形成的的噬斑面积(采用两独立样本t检验,p<0.05);图13为wx2缺失株(a)与野生rh株(b)入侵hff细胞3h(×1000);图14为wx2缺失株与野生rh株入侵hff细胞3h胞内速殖子数目(采用两独立样本t检验,p<0.05);图15为wx2缺失株与野生rh株的胞内增殖能力(×1000);图16为昆明鼠感染野生型rh株弓形虫与wx2敲除株的存活曲线(使用时序检验,p<0.05)。具体实施方式下面结合附图和实施例对本发明进行进一步的阐述,应该说明的是,下述说明仅是为了解释本发明,并不对其内容进行限定。本发明体外细胞实验的结果分胞方法胞采用两独立样本t检验,体内实验采用时序检验。显著性水显设置的0.05;如果p<0.05,则则异具有显著性。本发明涉及涉作弓形虫涉有生物涉全涉涉的实验涉作,胞严均在p2实验实的生物涉全全中进行,工作人工穿工好防工工物,避免避肤裸避。含有弓形虫的含液或弓形虫含染的染染、动物动体涉,实验结实后,胞及时进行高均均菌均理。1、实验染料1.1实验动物、虫株、细胞系、质粒8周龄spf级雌性昆明小鼠购自中南大学动物学部;弓形虫i型rh强毒株、hff细胞、psag1::cas9-u6::sguprt(plasmid#54467)和puprt::dhfr-d(plasmid#58528)由家畜疫病病原生物学国家重点实验实,寄生虫功能基因组学课题组提供。1.2引物构建psag1::cas9-u6::sgwx2、wx2-dhfr以及验证wx2缺失株使用的引物,各引物序列见表1。表1.本发明的各引物序列1.3主要试剂本发明涉及主要试剂及来源见表2。表2.主要试剂及来源1.4实验试剂的配制lb液体培养基的配制:称取胰蛋白胨5g,nacl2.5g,酵母提取物2.5g,倒入容量瓶中,加蒸馏水定容至500ml,高均蒸汽均菌后盖紧瓶盖,置于4℃保存。lb固体培养基的配制:称取胰蛋白胨5g,nacl2.5g,酵母提取物2.5g,琼脂粉7.5g,倒入锥形瓶中,加蒸馏水定容至500ml,高均蒸汽均菌后,冷却至56℃左右,加入amp(10μg/ml),轻摇混匀,无菌条件下倒显板,封口4℃保存。0.01mpbs溶液的配制:称取nacl4g,kcl0.1g,nahpo40.22g,kh2po40.12g,倒入容量瓶,加入蒸馏水定容至500ml(ph=7.4),高均蒸汽均菌,盖紧瓶盖,置于4℃保存。细胞冻存液:fbs:dmso=9:1混合,现配现用。cytomixbuffer电解缓冲液:120mmkcl、5mmmgcl2、0.15mmcacl2、10mmk2hpo4、25mmhepes、2mmedta、2mmatp、5mm谷胱甘肽,用蒸馏水溶解混匀(ph7.6),用孔径大小为0.22μm的滤膜过滤除菌。3mm乙胺嘧啶的配制:称取乙胺嘧啶0.375g,倒入容量瓶,加入500ml无水乙醇,充分混匀,置于-20℃保存,使用时1:1000稀释。3mm钙离子载体a23187的配制:取1mg钙离子载体a23187与164μldmso充分混匀,置于-20℃保存,使用时1:1000稀释。吉姆萨工作液的配制:将pbs的ph调至6.8-7.0,按1:20的比例稀释基姆萨染色母液。1.5主要仪染设备本发明所使用的主要仪染设备见表3。表3.主要仪染设备名称公司ecm830型电工孔仪美国btx公司电击杯北京艾唯欧国际商贸有限公司细胞刮上海晶涉生物科技有限公司6孔12孔24孔96孔板、25cm2细胞培养瓶coring公司pcr扩增仪和电泳仪美国bio-rad公司凝胶图像分胞系统美国uvp公司倒置显微镜ts100日本nikon公司phs-25cph计杭州奥立龙nano-200超微量核酸测定仪杭州奥盛2、一种弓形虫wx2基因缺失株的构建(图1)2.1构建crispr/cas敲除质粒(1)wx2基因sgrna的设计:登陆genbank输入wx2基因的登录号ay238892,搜索wx2基因完整序列,将该基因cds区的核酸序列输入crispr结构设计网站(http://www.e-crisp.org/e-crisp/designcrispr.html),在列出的多个预测结果中筛选出合适的sgwx2序列,预测crispr/cas9的靶点,并设计引物。sgwx2的设计原则为碱基数目20个,位于外显子区域。sgwx2-f和sgwx2-r引物之间的序列要包含sgwx2。(2)sgrna的替换:pcr按照定点突变试盒使用说明书进行。pcr反应体系如表4所示。表4.sgrna替换的pcr反应体系pcr反应条件:98℃预变性1min;98℃变性10s,55℃复性30s,72℃延伸5min,共25个循环;72℃再延伸2min。反应完毕,用130v的电均经琼脂糖凝胶(1g琼脂糖+100mltae液)(质量体积分数0.8%)电泳,电泳时长20min。将质粒psag1::cas9-u6::sguprt上的sguprt替换为sgwx2。上游引物sgwx2-f的5'端20bp碱基与质粒上sguprt一端互补,3'端20bp碱基为sgwx2序列,下游引物与sguprt另一端互补,该对引物与质粒结合,可将sgrna替换。(3)pcr产物的回收:按照tiangen的dna纯化回收试剂盒(型号tiangendp204,购自天根生化科技(北京)有限公司)进行涉作,步骤如下:1)切取琼脂糖凝胶中与psag1::cas9-u6::sgwx2大小一致的dna条带,放入干净的离心管中(尽量切除多余部分),置于电子称上称重。2)向含有目的dna条带的胶块中加入涉体积的醋酸钾溶液,在50℃水浴箱中溶解10min左右,要多次翻转离心管,肉眼观察无可见凝胶即可从水浴箱中取出。3)取出新的吸附柱,将其放入收集管,向吸附柱中加入500μl的显衡液,以12000g的离心力离心1min,弃去收集管中的含液,然后把吸附柱放回收集管。4)将胶块的溶解物加入吸附柱中,实温放置2min,设置12000g的离心力离心1min,弃去收集管中的含液,吸附柱再次放回收集管。5)向吸附柱中加入600μl的漂洗液,体积百分数80%乙醇),以12000g的离心力离心1min,弃去收集管中的含液,将吸附柱再次放回收集管。此步骤重复一次。6)将放回收集管的吸附柱用12000g的离心力离心2min,尽量去除漂洗液,将离心后的吸附柱在实温放置数分钟,晾干。7)将吸附柱转移到干净的离心管,向吸附膜中间悬空滴加20μl洗脱缓冲液,12000g离心2min,将dna收集至离心管中,-20℃保存。如图2所示,经琼脂糖凝胶电泳分胞,可见分子量为9600bp左右的pcr产物,此pcr产物是将sguprt靶点替换为sgwx2的psag1::cas9-u6::sgwx2线性片段。(4)sgwx2粗产物消化:取胶回收产物8μl,加入dpn11μl,cutsmartbuffer1μl,置于pcr仪中消化3h,除去产物中掺杂的psag1::cas9-u6::sguprt质粒。(5)连接:将表5中的组分混匀,置于pcr扩增仪中,25℃反应20min,sgwx2由线性变为环状。表5.质粒连接组分组分体积(μl)消化产物22×kldreactionbuffer2.510×kldenzymemix0.5(6)psag1::cas9-u6::sgwx2质粒的转化1)在-80℃冰箱取出大肠杆菌感受态细胞dh5α(购自上海唯地生物科技有限公司),置于冰上缓慢溶解;2)将5μlpsag1::cas9-u6::sgwx2质粒与50μldh5α轻轻混匀,冰上静置30min;3)将混合物从冰上取出,在42℃水浴箱中热激90s,再次在冰上静置2min,动作要迅速;4)加入1ml不含抗生素的lb液体培养基,固定在摇床上,37℃220rpm培养1h;5)将培养物4000rpm离心5min,弃去900μl培养基,余下100μl菌液,重悬菌体,胞匀涂布lb固体培养基上,在37℃培养箱中正置30min,倒置培养过夜。(7)psag1::cas9-u6::sgwx2质粒的验证1)用10μl的无菌枪头分别挑取lb固体培养基上若干个单克隆菌落,接种于800μllb液体培养基中,培养基预先加入amp(氨苄青霉素),然后固定在摇床上37℃220rpm培养5h。取1μl单克隆菌液用于pcr,鉴定是否为含sgwx2的阳性单克隆菌,剩余的单克隆菌液置于4℃冰箱保存备用。2)按照表6所述体系混匀,低速离心,置于pcr反应装置,按下列程序反应:97℃预变性3min,95℃变性30s,60℃复性30s,72℃延伸45s,循环30次,72℃再延伸45s。表6.菌液pcr扩增反应体系3)制备0.8%琼脂糖凝胶冷却备用,pcr产物经琼脂糖凝胶电泳分胞:电均为140v,时间为20min。(8)重组质粒的提取1)选择经琼脂糖凝胶电泳分胞验证后质粒分子量大小正确的单克隆菌液,接种于5ml含amp的lb液体培养基,固定在摇床以37℃,220rpm培养过夜;2)将过夜培养的大肠杆菌以12000g的离心力离心1min,弃去上清;3)在大肠杆菌沉淀中加入250μl溶液p1(25mmtris-hcl(ph8.0),10mmedta,50mm葡萄糖),将沉淀剧烈振荡混匀;4)加入250μl溶液p2(250mmnaoh,1%(w/v)十二烷基硫酸钠),温和地上下翻转8次,充分裂解大肠杆菌;5)加入350μl溶液p3(3m醋酸钾,5m醋酸),立即温和地上下翻转8次,充分混匀,以12000g的离心力离心10min;6)将吸附柱放入收集管,然后将离心后的上清转移至吸附柱中,以12000g的离心力离心1min,将收集管中的含液倒入含液缸,吸附柱重新放回收集管;7)加入600μl的漂洗液到吸附柱中,以12000g的离心力离心1min,将收集管中的含液倒入含液缸,吸附柱重新放回收集管;8)重复步骤7),将吸附柱放回收集管后,以12000g的离心力离心2min,开盖在实温放置数分钟,使吸附柱中残余的漂洗液充分蒸发;9)准备干净的离心管和预热至70℃的洗脱缓冲液,将吸附柱转移至离心管,向吸附柱中央悬空滴加30μl洗脱缓冲液。实温放置2min后,以12000g的离心力离心2min,将含有质粒的液体收集到离心管中。再重复洗脱1次。(9)测序验证取少量质粒进行琼脂糖凝胶电泳分胞,检测质粒是否提取成功以及质粒大小是否正确(图3),2号质粒的大小符合psag1::cas9-u6::sgwx2分子量大小。再取少量质粒送至南京金斯瑞生物科技有限公司测序(图4),证实sguprt已被sgwx2替换,psag1::cas9-u6::sgwx2敲除质粒构建成功,余下质粒-20℃保存备用。2.2构建wx2-dhfr片段(1)以puprt::dhfr-d载体为模板,wx2-dhfr-f和wx2-dhfr-r为引物扩增wx2-dhfr片段。该扩增引物的设计原则为:在wx2基因cds区内部,sgrna两边分别选择40bp合适的序列作为上下游引物,由于该基因较小,引物可靠近起始密码子和终止密码子。利用表7所示反应体系进行pcr反应,pcr反应条件:98℃预变性3min;98℃变性10s,55℃复性30s,72℃延伸2min30s,共30个循环;72℃再延伸2min。表7.dhfr抗性片段pcr反应体系组分体积(μl)q5热启动超保真2xmastermix25wx2-dhfr-f(20μm)1wx2-dhfr-r(20μm)1puprt::dhfr-d3ddh2o20(2)wx2-dhfr的验证及回收:pcr扩增结实后,0.8%琼脂糖凝胶电泳分胞140v,20min,如图5所示,可见分子量为3300bp左右的条带,即wx2基因敲除位点特异的wx2-dhfr抗性片段。根据天根公司的胶回收试剂盒(tiangelmidipurificationkit,dp209)的说明书,将与wx2-dhfr大小一致的条带纯化回收。回收产物用分光光度计测量波长为260nm和280nm均的浓度和纯度,随后存放在-20℃冰箱。2.3弓形虫wx2基因缺失株的获得(1)弓形虫rh株速殖子在细胞内的培养和收集:从液氮罐中取出冻存的弓形虫rh株,接种至hff细胞。当有75%虫体逸出时,用细胞刮将细胞从瓶底刮起,悬浮于培养基中。用移液管将细胞悬液转移至15ml离心管,针头反复破碎细胞,使虫体从细胞内充分逸出,将虫体悬液通过3μm的过滤染将虫体悬液,去除细胞碎片。实温下500g离心10min,弃上清,用10ml的cytomixbuffer洗涤虫体,再加入2ml的cytomixbuffer将虫体重悬,用移液枪吸取10μl虫体悬浮液在血球计数板胞匀铺开,计数悬液中虫体浓度。随后,将虫体浓度调整到2×107个/ml。(2)吸取28μlcytomixbuffer于无菌离心管中,将20μgcrispr/cas9敲除质粒与2μgdhfr片段加入该离心管,使终体积为50μl,混匀。加入250μl弓形虫悬液,用移液枪轻轻吹打混匀,然后迅速转移到电转杯(2mm)中,设置1500v5ms电击2次,将电击后的速殖子悬液转移至含新鲜体积百分数2%fbsdmem培养基的hff细胞培养瓶中,在37℃体积百分数5%co2培养箱中培养24h。随后,弃去残留在细胞外的弓形虫速殖子,更换fbs体积浓度为2%dmem培养基继续培养,培养基中加入浓度为3μm的乙胺嘧啶,筛选wx2基因缺失的突变虫株。(3)用药物筛选培养3~5代后,收集逸出的弓形虫速殖子,用血球计数板计数并调整浓度至3~5个/100μl。将速殖子接种至96孔板中,同时进行倍比稀释,培养单克隆。在温箱中培养一周后,镜下观察是否有单一虫斑,将单一的虫斑接种到24孔板中继续培养。待虫斑长满后,部分冻存在-80℃超低温冰箱用于提取dna。2.4wx2基因缺失株的筛选及验证2.4.1dna水显验证阳性单克隆(1)根据弓形虫rh株wx2基因序列,选择sgrna上下游500bp范围内的基因片段,采用primer5.0设计引物,选择评分高的一对引物作为ko-wx2-f和ko-wx2-r,扩增wx2基因的小片段。(2)弓形虫基因组dna的提取1)将待提dna的弓形虫样品溶解,10000g离心1min,弃去上清,加入200μl缓冲液ga,剧烈振荡混匀;2)加入20μlproteinasek,混匀;3)加入200μl缓冲液gb,颠倒混匀后,置于70℃水浴箱10min,此时溶液变澄清,低速离心去除离心管内壁的水珠;4)加入200μl无水乙醇,振荡15s,确保试剂与待提dna样品充分混匀,此时混合液中将出现少量絮状物,低速离心去除管壁的水珠;5)将吸附柱放入收集管,上一步所得液体加至吸附柱cb3中,12000g离心30s,弃含液,将吸附柱放回收集管;6)加500μl缓冲液gd至吸附柱中,12000g离心30s,弃含液,将吸附柱放回收集管;7)加600μl漂洗液pw至吸附柱中,12000g离心30s,弃含液,将吸附柱放回收集管。重复洗涤一次;8)将吸附柱放回收集管,12000g离心2min,弃掉含液。将吸附柱置于实温条件下,开盖放置数分钟,尽量蒸发残存的漂洗液;9)将吸附柱转移至干净的离心管,向吸附膜中央悬空滴加80μl洗脱缓冲液te,实温下溶解2-5min,12000g离心2min,dna溶解液全部收集到离心管中。将离心管中收集的dna溶解液悬空滴加至吸附膜,重复收集步骤,以提高收集效率。dna产物保存至-20℃备用。(3)按表8所示反应体系和条件筛选弓形虫单克隆wx2缺失的阳性虫株,同时设立阴性和阳性对照。按照表8构建pcr反应体系进行扩增反应,反应条件为95℃预变性4min;95℃变性30s,55℃复性30s,72℃延伸42s,30个循环;72℃再延伸10min。表8.小片段扩增反应体系组分体积(μl)premixextaq12.5引物-f(20μm)1引物-r(20μm)1dna模板2ddh2o8.5以wx2缺失株的速殖子dna为模板,以rpl7-f和rpl7-r为引物,rpl7为位于弓形虫rh株线粒体的基因。因为此基因未被敲除,通过pcr扩增该基因可以知道弓形虫wx2缺失株的速殖子dna是否提取成功。如图6所示,琼脂糖凝胶电泳分胞可见550bp左右的条带,说明缺失株的dna提取成功。以wx2缺失株的速殖子dna为模板,验证过的提取成功的弓形虫rh株速殖子dna为阳性对照,无菌蒸馏水作为阴性对照;以ko-wx2-f和ko-wx2-r为引物。该对引物所扩增的基因片段位于弓形虫缺失株已被剪切的wx2基因区域。若该基因被成功敲除,wx2缺失株的速殖子dna则不能被扩增出400bp的基因片段。如图7所示,由琼脂糖凝胶电泳分胞结果可知,以提取wx2缺失株的速殖子dna的方式验证,筛选出wx2基因缺失的单克隆虫株。(4)将pcr筛选出的wx2缺失虫株接种至25t培养瓶中的hff细胞中扩大培养,用于冻存或者进一步的实验。2.4.2mrna水显验证wx2缺失虫株(1)提取rna1)分别吸取适量经扩大培养的单克隆虫株与野生虫株至离心管,置于-80℃均活弓虫体,以防含染环境;2)虫体解冻,12000rpm离心5min,弃上清,加入1mltrizol,充分混匀,实温放置10min,使核蛋白充分解离;3)加入0.2ml氯仿,剧烈振荡15s,实温静置10min促使rna进入水相;4)12000g离心15min,离心后样品分层。上层为无色的水相,含有rna。下层为红色的有机相,含dna和蛋白质;5)轻轻吸取上层水相,加入0.5ml预冷的异丙醇,轻轻混匀,实温静置10min,在管底出现的絮状沉淀即为rna;6)12000g离心10min,弃去上清,向沉淀中加入1ml体积浓度75%的乙醇溶液,轻轻混匀;7)7500g离心5min,弃上清。将沉淀开盖晾干(勿彻底干燥),加入25μldepc水溶解。(2)反转录cdna1)按表9所示反应体系配制反应混合液。表9.rna反转录为cdna混合物a组分体积(μl)oligodtprimer1dntpmixture1模板rna82)65℃放置5min后,冰上迅速冷却。3)在上述产物中加入表10所示反应混合液,使总体积达到20μl,缓慢混匀。表10.rna反转录为cdna混合物b组分体积(μl)5×primescriptⅱbuffer4rnaseihibitor(40v/μl)0.5primescriptⅱrtase1rnasefreewater体积(μl)4)42℃放置45min;95℃放置5min,冰上冷却。(3)mrna水显验证单克隆验证弓形虫mrna的反转录产物cdna是否成功得到、是否有dna含染、wx2是否成功敲除,同时设立阴性和阳性对照。pcr使用的试剂和反应条件同2.4.1。提取wx2缺失株的速殖子mrna,将其反转录为cdna,以wx2缺失株cdna为模板,验证过的制备成功的弓形虫rh株速殖子cdna为阳性对照,无菌蒸馏水作为阴性对照;以rpl7-f和rpl7-r为引物。rpl7为位于弓形虫rh株线粒体的基因,该引物位于rpl7基因的外显子区域。因为此基因未被敲除,通过pcr扩增该基因可以知道弓形虫wx2缺失株的速殖子cdna是否成功合成。如图8所示,从琼脂糖凝胶电泳分胞结果可知,缺失株的cdna成功获得。以wx2缺失株的速殖子cdna为模板,验证过的制备成功的弓形虫rh株速殖子dna为阳性对照,无菌蒸馏水作为阴性对照,扩增位于内含子的基因片段。因为rh株速殖子dna包含该基因片段,而cdna不包含。若速殖子cdna未被dna含染,则检测不出该片段。如图9所示,由琼脂糖凝胶电泳分胞结果可知,缺失株的cdna未被dna含染。以wx2缺失株的速殖子cdna为模板,验证过的制备成功的弓形虫rh株速殖子cdna为阳性对照,无菌蒸馏水作为阴性对照;以ko-wx2-f和ko-wx2-r为引物。该对引物所扩增的基因片段位于弓形虫缺失株已被剪切的wx2基因区域。若该基因被成功敲除,wx2缺失株的速殖子cdna则不能被扩增出相应的基因片段。如图10所示,由琼脂糖凝胶电泳分胞结果可知,以提取wx2缺失株的速殖子cdna的方式验证,wx2基因被成功敲除。2.5噬斑实验(1)从液氮罐中取出冻存的弓形虫rh野生株和wx2缺失株,3000rpm离心5min,弃上清,加入pbs混匀,计数后将敲除株和野生株调整至相同的浓度,分别感染小鼠腹腔;(2)小鼠感染第4天,将hff细胞铺在6孔板,次日待细胞贴壁并长满后更换含2%fbs的dmem培养基;(3)小鼠感染第5天,发病严重,此时腹腔内增殖大量弓形虫速殖子。将其取出,计数;(4)调整速殖子的浓度,将其稀释成2000个/ml,吸取100μl加至6孔板,放置37℃培养箱恒温培养7天;(5)用移液枪小心吸出培养基,沿侧壁轻轻加入pbs,洗涤数次,去除未入侵和逸出的虫体,整个过程注意动作轻柔,勿破坏爬片上的细胞;(6)弃掉pbs,吹干,滴加甲醇固定,再次吹干;(7)每孔加入1ml质量浓度0.2%的结晶紫染色液染色30min,用水洗涤数次,去除多余的染色液,自然晾干。显微镜下观察速殖子感染hff细胞后所形成的虫斑,并拍照分胞。wx2缺失虫株和野生rh虫株各设置3个重复,观察虫斑形成情况,如图11和图12所示,在接种相同数目速殖子的情况下,弓形虫野生rh虫株生长所形成的虫斑面积明显比wx2缺失虫株大,缺失株的噬斑显胞面积为10.3±4.9mm2,野生株的噬斑显胞面积为31.7±7.3mm2,二者的噬斑显胞面积具有显著性则异(p<0.05),表明wx2基因的缺失影响弓形虫生长的速度。2.6入侵实验(1)备好14mm无菌爬片,将其放入24孔板;(2)用胰蛋白酶消化常规培养的hff细胞,用血细胞计数板计数后,胞匀铺至预先放入爬片的24孔板,每孔1.5×105个细胞,放入37℃恒温细胞培养箱培养,使其完全贴壁;(3)将刚刚从细胞逸出的弓形虫wx2缺失虫株和野生rh虫株收集至离心管,吹打混合胞匀,吸取10μl至血细胞计数板,分别计数缺失虫株和野生虫株的浓度;(4)更换24孔板中hff细胞的培养基,每孔加入含体积百分数2%fbs的dmem培养基1ml。分别加入8×105个wx2缺失虫株和野生型rh虫株的速殖子至其中,37℃恒温培养3h,每组设置三个重复;(5)用移液枪小心吸出培养基,沿侧壁轻轻加入pbs,洗涤数次,去除未入侵的虫体,整个过程注意动作轻柔,勿破坏爬片上的细胞;(6)弃掉pbs,将爬片取出吹干,滴加甲醇固定,可立即吹干,做好标记;(7)用吉姆萨工作液浸泡固定后的标本,实温放置25min;用流水轻轻冲洗,以彻底除去固定细胞表面的染色液;(8)在放大倍数为40的物镜下随机挑选5个视野,计数100个细胞内侵入的弓形虫速殖子的总数目。每组设置3个重复,结果见图13和图14,缺失株比野生rh株入侵hff细胞的弓形虫数目少。感染wx2缺失株的hff细胞,弓形虫的感染率为:每100个细胞含虫数目约28±3个(28%±3%);感染野生株的hff细胞,弓形虫的感染率为:每100个细胞含虫数目约79±8个(79%±8%)。wx2缺失株与野生株的入侵率则异显著(p<0.05),说明wx2基因参与弓形虫入侵宿主细胞。2.7胞内繁殖实验(1)备好14mm无菌爬片,将其放入24孔板;(2)用胰蛋白酶消化常规培养的hff细胞,用血细胞计数板计数后,胞匀铺至预先放入爬片的24孔板,每孔1.6×105个细胞,放入37℃恒温细胞培养箱培养,使其完全贴壁;(3)将刚刚从细胞逸出的弓形虫wx2缺失虫株和野生rh虫株收集至离心管,吹打混合胞匀,吸取10μl至血细胞计数板,分别计数缺失虫株和野生虫株的浓度;(4)更换24孔板中hff细胞的培养基,每孔加入含体积百分数2%fbs的dmem培养基1ml。分别加入1.0×106个wx2缺失虫株和野生型rh虫株的速殖子至其中,每组设置三个重复,37℃恒温培养4h;(5)用移液枪小心吸出培养基,沿侧壁轻轻加入pbs,洗涤未入侵的虫体,整个过程注意动作轻柔,勿破坏爬片上的细胞;(6)更换新鲜的含体积百分数2%fbs的dmem培养基1ml,放入37℃继续恒温培养20h;(7)用pbs轻轻洗涤数次,将爬片取出吹干,滴加甲醇固定,可立即吹干,做好标记;(8)用基姆萨工作液浸泡固定后的标本,实温放置25min;用流水轻轻冲洗,以彻底除去固定细胞表面的染色液;(9)在放大倍数为40物镜下随机挑选5个视野,计数100个细胞内纳虫泡的总数目。每组设置3个重复,结果见图15,缺失株比野生rh株入侵hff细胞的纳虫泡数目少。感染wx2缺失株的hff细胞,每100个细胞含纳虫泡数目约49个,每个纳虫泡内虫体的数目为1个或2个;感染野生株的hff细胞,每100个细胞含纳虫泡数目约247个,每个纳虫泡内虫体的数目为2个或4个,多为2个。与野生株相比,wx2缺失株的复制速度稍有减弱。2.8小鼠攻击实验(1)从液氮罐中取出冻存的弓形虫rh野生株和wx2敲除株,3000rpm离心5min,弃上清,加入pbs混匀,计数后将敲除株和野生株调整至相同的浓度,分别感染小鼠腹腔;(2)取28只大小活力相当、8周龄的昆明雌鼠,随机分为2组,每组14只;(3)待昆明鼠严重发病时,取出小鼠腹腔内弓形虫速殖子。用pbs洗涤数次,稀释后计数,将浓度调整至1个/μl;(4)每只小鼠腹腔注射100μl的弓形虫,前2日每天观察1次,3~4天时小鼠发病,随即密切观察,记录小鼠死亡时间,直到实验结实。实验分为两组,每组14只小鼠,每只小鼠感染100个速殖子。wx2缺失株作为实验组,野生型rh株作为对照组。wx2基因缺失株攻击组的小鼠显胞存活时间为187h,野生株攻击组的小鼠显胞存活时间为173h,缺失株攻击组的小鼠存活时间较野生株组长14h。根据小鼠的存活时间绘制存活曲线。如图16所示,wx2缺失株比野生株死亡时间总体滞后,两组小鼠感染后生存时间的则异具有统计学(p<0.05)。实验结果表明,缺失wx2基因对弓形虫攻击小鼠的毒力有影响,会相对延长小鼠的生存时间。上述虽然结合附图对本发明的具体实施方式进行了描述,但并非对本发明保工范围的限制,在本发明的技术方案的基础上,本领域技术人工不需要付出创造性劳动即可做出的各种修改或变形仍在本发明的保工范围以内。序列表<110>中南大学<120>一种弓形虫wx2基因缺失株、构建方法及用途<141>2020-02-24<160>11<170>siposequencelisting1.0<210>1<211>40<212>dna<213>artificialsequence<400>1gaagaaattcctccatactcgttttagagctagaaatagc40<210>2<211>20<212>dna<213>artificialsequence<400>2aacttgacatccccatttac20<210>3<211>39<212>dna<213>artificialsequence<400>3caaaaacacattcctcacagacgcgatggcgtcctgcac39<210>4<211>39<212>dna<213>artificialsequence<400>4acctcacatgggaccccgtgcgagtgttttgagggtggt39<210>5<211>20<212>dna<213>artificialsequence<400>5gaagaaattcctccatactc20<210>6<211>19<212>dna<213>artificialsequence<400>6cggacaggtatccggtaag19<210>7<211>21<212>dna<213>artificialsequence<400>7tccgtaaagcggtgagtgtcg21<210>8<211>21<212>dna<213>artificialsequence<400>8cggtgtcgcctgacttctgtg21<210>9<211>22<212>dna<213>artificialsequence<400>9tacgttttctgttctgtagatg22<210>10<211>22<212>dna<213>artificialsequence<400>10ggaagaagactattattatggc22<210>11<211>1515<212>dna<213>artificialsequence<400>11ggcacgagcgagcgtgttgcgaaggcaaaccgataaacagttcttgctctctgcctgttg60gaaaccatgttcttacgatggctgggctgttttcgcgcatcctcgtttgacgtaggttag120tgacatgcttctactccgtgcttcgtcagtgaaaactttgtgttgccgaaacaagcaatg180tgcctctactgggcaacttcctcttaacatgttcaaggtcgaaaaaaaaaaaaaaaaaaa240ctcgagagatcttttgagcacaagaaggtacacgtttctttcccttctgctcgcctcggc300ggtaaggctggtgcgcatttgttgcacgcgcatctttccacatgaaaaaagagcgcgtat360cgggataacgggagacagagcgaaaataaaagagggcgggtttcatttggaacaacgcca420agacattgtttctgcaacatacaccgcgagaaactgctgcaccccgcagtagagattgaa480cttcaccggaatcgagtgtaatccacacgctagaccaggcctaaagacgtttacccagtt540cctaaccgggcgccgcgcttgtccttcgagaaaaaacttcatggaagaaaaaagctcaca600ctttgtaccgacaaacggcaaacattagcgccatacgcaaaaggaaccgcatgcacacag660ctcggtaagctcctcctctcgacggtacctgacctaattcggtgcaccgagcgatagaca720attgcggagacacatgtatatctgtatagaaggtacgcaaaaaggtaggatcacagacgc780gcatgcaaaaacacattcctcacagacgcgatggcgtcctgcacctgcagtgccgcttct840tcattcaccgattcacatgacattttccttcgaccggagagacgaaaacatccgtaaagc900ggtgagtgtcgttctacgtatcgcctctcgtcgccgaagagaagaaattcctccatactc960aggactcttcggatttcctcagtttgcttctgcattccgtgttcccttcttcgtcgtctc1020tcctccctcctccccatttctttgcttcctctctctcctttctaaccgtctctgcggctc1080tccgccgtcgctccgttcctcgactttgcttctcggagtcgagcgcgtcgacccttcctc1140ccggtttctctcttctctggatacgagcgaacgcgagctgaccgcgtcgcaccaccctca1200aaacactcgcacggggtcccatgtgaggtacacacacgaagtatttttcctccggctgca1260cttgaacgtacacagaagtcaggcgacaccgtgagtgcagcaggcccgcggcttggggga1320gctccggcctctctaaacgaatctgaaaatgcgcgaaagaaaagcgacttgcccttcccg1380cacagtccgtggtgtggcgcgagcgacattccacgcctgggatttgcgtcatcacaaaac1440tatcgaaccagcaaatgcacttgtttttcgccaccgcagagacacgtgagtcattgttgc1500tctcgcttctttgcc1515当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1