单环倍半萜类化合物及其用途
:
1.本发明属于医药技术领域,涉及单环倍半萜类化合物及其用途,具体涉及从药用植物灯盏细辛全草中分离得到的8个具有相同平面结构的单环倍半萜类化合物(四对对映异构体),及其在制备抗血栓药物中的应用。
背景技术:
2.灯盏细辛,又名灯盏花、灯盏草、短茎飞蓬,为菊科植物短葶飞蓬erigeron breviscapus(vant.)hand.-mazz.的干燥全草,多见于山地疏林下、草丛或向阳坡地。主要分布于我国云南、广西、四川、贵州和西藏等西南部地区,其性寒,味辛、微苦、温,归心、肝经,全草入药,具有散热解表、舒经治瘫、活血通络止痛、祛风散寒的功效,已收入2015年版《中国药典》。现代药理研究表明,灯盏细辛具有心脑血管保护、神经保护、抗氧化、抗癌等药理作用。
3.灯盏细辛为传统中药,主要用于治疗脑中风及其后遗症。自20世纪80年代,已开发了片剂和注射剂在临床上用于治疗脑栓塞、脑血栓形成、脑血栓引起的瘫痪、冠心病、心绞痛、急性肾功能衰退等,在临床上有非常确切的疗效。目前研究中,灯盏乙素(野黄芩苷)及二咖啡酰基奎宁酸酯类化合物是其中的主要有效成分。萜类天然产物结构的多样性决定了其生物活性多样性,我们不难从中发现一些研究报道关于萜类成分在心血管疾病中所发挥的重要作用。最有效的萜类包括潜在的高血压治疗药物citronellol,linalool,预防血栓形成的(+)-nootkatone和石竹烯氧化物,以及防止或改善心肌梗死的triptolide。一些临床试验也提供了萜类成分如sterol glycosides,ginsenosides and carotenoid等在心血管医药领域发挥的巨大潜力。但对于该类化合物的研究还很有限,有待于更进一步地研究。
技术实现要素:
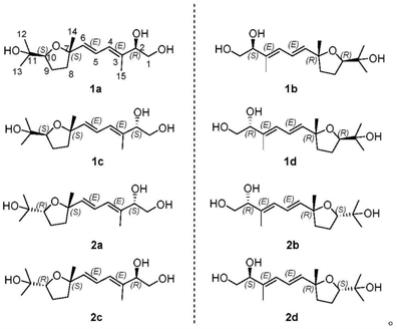
4.为了克服现有技术的缺陷,本发明提供了一系列单环倍半萜类化合物、异构体或其盐,其结构如下:
[0005][0006]
进一步地,本发明提供了如下的单环倍半萜类化合物、异构体或其盐:
[0007][0008]
所述的单环倍半萜类化合物、异构体或其盐是从菊科飞蓬属植物灯盏细辛(erigeron breviscapus(vant.)hand.-mazz.)中同时分离得到的。
[0009]
本发明提供了所述的单环倍半萜类化合物、异构体或其盐的制备方法,包括如下步骤:
[0010]
(1)灯盏细辛干燥全草以乙醇加热回流提取,合并提取液,浓缩得浸膏,浸膏采用二氯甲烷或三氯甲烷进行萃取得到二氯甲烷或三氯甲烷层及水层浸膏;
[0011]
(2)将水层浸膏经大孔吸附树脂柱使用乙醇-水系统(50%-80%)进行快速梯度洗脱,经硅胶薄层检识分为三个馏分a、b、c;
[0012]
(3)馏分c再经快速减压硅胶柱色谱以二氯甲烷/三氯甲烷-甲醇-水系统(9.0:1.0:0.0-1.0:1.0:0.8)进行快速梯度洗脱,经硅胶薄层检识进行合并后共得三个馏分c1、c2、c3;
[0013]
(4)馏分c1经聚酰胺柱色谱,以乙醇-水系统(60%-90%)进行梯度洗脱,共收集到三个组分c
1-1
、c
1-2
、c
1-3
。
[0014]
(5)所得组分c
1-1
经凝胶柱色谱以乙醇-水(60:40)进行等度洗脱,得到三个组c
1-1-1
、c
1-1-2
、c
1-1-3
。
[0015]
(6)组分c
1-1-2
经硅胶柱色谱以二氯甲烷/三氯甲烷-甲醇系统(50:1-1:1)进行梯度洗脱,经薄层检识和hplc分析进行合并得到五个组分c
1-1-2-1-c
1-1-2-5
。
[0016]
(7)所得组分c
1-1-2-2
经ods柱色谱以甲醇-水(20%-90%)进行梯度洗脱,得到六个组分c
1-1-2-2-1-c
1-1-2-2-6
。
[0017]
(8)利用半制备hplc对组分c
1-1-2-2-5
以25%乙腈水系统进行洗脱,得到了一对差向异构体1和2,其平面结构如下:
[0018][0019]
其中,步骤(1)中所述的乙醇为70-80%工业乙醇,提取2-3次。二氯甲烷或三氯甲烷与浸膏的体积比例为1:1,萃取3-4次。
[0020]
进一步地,
[0021]
对这对差向异构体1和2进行手性拆分得到化合物1a-1d和2a-2d。其手性制备拆分方法为:利用daicel chiralpak ad-h和daicel chiralpak ic手性色谱柱,流动相为正己烷-异丙醇混合溶剂,比例分别为8:1和5:1(v:v),流速分别为2.00和0.45ml/min,紫外检测器检测波长为210nm。
[0022]
本发明中所述的手性拆分条件,适用于化合物1和2。
[0023]
本发明所得化合物的结构鉴定结果如下:
[0024]
利用高分辨质谱、一维和二维nmr技术对化合物1、2的平面结构及相对构型进行确定。通过比较实测ecd和计算ecd谱图,mosher's法对拆分后的光学纯化合物1a-1d和2a-2d的绝对构型进行确定。
[0025]
化合物1:透明油状化合物,hresi-ms给出准分子离子峰m/z 293.1769[m+na]
+
(calcd for c
15
h
26
o4,293.1723),结合1h,
13
c nmr数据确定其分子式为c
15
h
26
o4,不饱和度为3。1h nmr(600mhz,cdcl3)谱中,低场区可见δ
h 6.41(1h,dd,j=15.2,10.9,1.8hz),6.12(1h,d,j=10.9hz),5.74(1h,d,j=15.2hz)为相互偶合烯氢信号,δ
h 4.17(1h,dd,j=7.8,3.5hz),3.80(1h,t,j=7.2hz)为两个连氧次甲基氢信号,δ
h 3.65(1h,dd,j=11.2,3.9hz),3.54(1h,dd,j=11.2,7.8hz)为一个连氧亚甲基氢信号。高场区可见4个甲基氢信号δ
h 1.77(3h,m),1.33(3h,s),1.22(3h,s),1.13(3h,s),其余δ
h 1.90(1h,m),1.77(1h,m)和1.83(2h,m)为脂肪族氢信号。
13
c nmr(100mhz,cdcl3)和hsqc谱推测含有15个碳信号,提示化合物1为倍半萜。其中包括两组共轭的双键信号δ
c 140.0,135.9,125.7,122.3,五个连氧碳信号δ
c 85.8,83.0,77.0,71.4,65.4,六个sp3杂化碳δ
c 38.2,27.3,27.3,26.6,24.3,13.6。结合hsqc,hmbc谱对该化合物做进一步解析。
[0026]
通过hsqc谱对直接相连的碳氢信号进行了归属。在hmbc谱中,h-8(δ
h
1.90)与c-6(δ
c 140.0),c-10(δ
c 85.8)存在远程相关;h-9(δ
h 1.83)与c-7(δ
c 83.0),c-11(δ
c 71.4)存在远程相关,推测存在一个呋喃环。h-1(δ
h 3.54)与c-2(δ
c 77.0),c-3(δ
c 135.9)存在远程相关;h-2(δ
h 4.17)与c-1(δ
c 65.4),c-4(δ
c 125.7)存在远程相关;h-4(δ
h 6.12)与c-5(δ
c 122.3),c-6(δ
c 140.0)存在远程相关;h-6(δ
h 5.74)与c-7(δ
c 83.0),c-8(δ
c 38.2)存在远程相关;h-5(δ
h 6.41)与c-7(δ
c 83.0)存在远程相关。h
3-12(δ
h 1.13)与c-10(δ
c 85.8),c-11(δ
c 71.4),c-13(δ
c 27.3)存在远程相关;h
3-13(δ
h 1.22)与c-10(δ
c 85.8),c-11(δ
c 71.4),c-12(δ
c 24.3)存在远程相关;h
3-14(δ
h 1.33)与c-6(δ
c 140.0),c-7(δ
c 83.0),c-8(δ
c 38.2)存在远程相关;h
3-15(δ
h 1.77)与c-2(δ
c 77.0),c-3(δ
c 135.9),c-4(δ
c 125.7),c-5(δ
c 122.3)存在远程相关,推测四个甲基的连接位置。以上相关共同推测化合物1的平面结构。
[0027]
通过noesy谱和偶合常数确定了化合物1的相对构型。在noesy谱中,h-10(δ
h 3.80)与h
3-14(δ
h 1.33)没有noesy相关,表明h-10和h
3-14不共平面,处于呋喃环的异侧;
h
3-15(δ
h 1.77)与h-1(δ
h 3.54),h-5(δ
h 6.41)有相关,推测
△
3,4
双键的构型为e构型。另外,h-5和h-6的j
5,6
=15.2hz证明
△
5,6
双键的构型为e构型。
[0028]
化合物2:透明油状,hresi-ms给出准分子离子峰m/z 293.1724[m+na]
+
(calcd for c
15
h
26
o4,293.1723),结合1h,
13
c nmr数据确定其分子式为c
15
h
26
o4,不饱和度为3。1h nmr(600mhz,cdcl3)谱中,低场区可见δ
h 6.43(1h,dd,j=15.4,10.9hz),6.12(1h,d,j=10.9hz),5.83(1h,d,j=15.4hz)为相互偶合烯氢信号。此外,δ
h 4.17(1h,dd,j=7.7,3.5hz),3.65(1h,dd,j=11.2,3.6hz),3.54(1h,dd,j=11.2,7.6hz),3.86(1h,t,j=7.2hz)为3个连氧碳氢信号。高场区可见δ
h 1.76(3h,s),1.35(3h,s),1.23(3h,s),1.13(3h,s)为四个甲基氢信号,其余δ
h
1.92(1h,m),1.82(1h,m)和1.87(2h,m)为脂肪族氢信号。
13
c nmr(100mhz,cdcl3)和hsqc谱推测含有15个碳信号,提示化合物2为倍半萜。其中包括两组共轭的双键信号δ
c 140.2,136.4,125.7,122.7,五个连氧碳信号δ
c 85.7,82.8,77.1,71.5,65.4,六个sp3杂化碳信号δ
c 38.6,27.5,26.8,26.5,24.5,13.6为sp3杂化碳。结合hsqc及hmbc谱对该化合物做进一步解析。
[0029]
通过hsqc谱对直接相连的碳氢信号进行了归属。在hmbc谱中,h-8(δ
h
1.92)与c-6(δ
c 140.2),c-10(δ
c 85.7)存在远程相关;h-9(δ
h 1.87)与c-7(δ
c 65.4),c-11(δ
c 125.7)存在远程相关,推测存在一个呋喃环。h-1(δ
h 3.54)与c-2(δ
c 77.1),c-3(δ
c 136.4)存在远程相关;h-2(δ
h 4.17)与c-1(δ
c 65.4),c-4(δ
c 125.7)存在远程相关;h-4(δ
h 6.12)与c-5(δ
c 122.7),c-6(δ
c 140.2)存在远程相关;h-5(δ
h 6.43)与c-7(δ
c 82.8)存在远程相关;h-6(δ
h 5.83)与c-7(δ
c 82.8),c-8(δ
c 38.6)存在远程相关。h
3-12(δ
h 1.13)与c-10(δ
c 85.7),c-11(δ
c 71.5),c-13(δ
c 27.5)存在远程相关;h
3-13(δ
h 1.23)与c-10(δ
c 85.7),c-11(δ
c 71.5),c-12(δ
c 24.5)存在远程相关;h
3-14(δ
h 1.35)与c-6(δ
c 140.2),c-7(δ
c 82.8),c-8(δ
c 38.6)存在远程相关;h
3-15(δ
h 1.76)与c-2(δ
c 77.1),c-3(δ
c 136.4),c-4(δ
c 125.7),c-5(δ
c 122.7)存在远程相关,推测四个甲基的连接位置。以上相关共同推测化合物2的平面结构。
[0030]
通过noesy谱确定了化合物2的相对构型,如图所示(fig.2-4),h-10(δ
h 3.86)与h
3-14(δ
h 1.35)有noesy相关,表明h-10和h
3-14共平面,处于呋喃环的同侧。另外分别根据noest谱和偶合常数确定
△
3,4
和
△
5,6
双键的相对构型,h
3-15(δ
h 1.76)与h-1(δ
h 3.54),h-5(δ
h 6.43)有相关,推测
△
3,4
双键的构型为e构型;h-5和h-6的j
5,6
=15.4hz证明
△
5,6
双键的构型为e构型。
[0031]
化合物1和2的比旋光度接近于零,且无明显的cd吸收,推测其为外消旋混合物。使用daicel chiralpak ad-h和daicel chiralpak ic手性色谱柱对化合物1和2进行进一步的分离,结果得到四对对映异构体1a-1d和2a-2d。它们的绝对构型是通过比较计算和实测ecd来确定的。
[0032]
表1 1-2在cdcl3中1h(600mhz)与
13
c(100mhz)nmr数据
[0033][0034]
附图说明:
[0035]
图1化合物1的hresims谱;
[0036]
图2化合物1的hmbc谱(600mhz,cdcl3);
[0037]
图3化合物1的hsqc谱(600mhz,cdcl3);
[0038]
图4化合物1的noesy谱(600mhz,cdcl3);
[0039]
图5化合物2的hresims谱;
[0040]
图6化合物2的hmbc谱(600mhz,cdcl3);
[0041]
图7化合物2的hsqc谱(600mhz,cdcl3);
[0042]
图8化合物2的noesy谱(600mhz,cdcl3);
[0043]
图9化合物1和2的手性拆分色谱图;
[0044]
图10化合物1a-1d和2a-2d实测ecd。
具体实施方式:
[0045]
下面所列实施例有助于本领域技术人员更好地理解本发明,但不以任何方式限制本发明。
[0046]
实施例1:化合物1-2的制备。
[0047]
选用20kg菊科植物短葶飞蓬erigeron breviscapus(vant.)hand.-mazz.的干燥全草为原料,用80%工业乙醇加热回流提取3次,每次2小时。经减压浓缩后得乙醇粗提物4kg,浸膏采用二氯甲烷以1:1的体积比进行萃取,萃取3次,得到二氯甲烷层600g及水层浸膏3.2kg。将水层浸膏经大孔吸附树脂柱使用50%-80%乙醇-水系统进行快速梯度洗脱,经硅胶薄层检识合并为三个馏分a(1.1kg)、b(1.1kg)、c(0.9kg)。馏分c经快速减压硅胶柱色谱,以二氯甲烷-甲醇-水系统(9.0:1.0:0.0-1.0:1.0:0.8)进行快速梯度洗脱,经硅胶薄层检识进行合并后共得三个馏分c1(210g)、c2(243g)、c3(186g)。馏分c1经聚酰胺柱色谱,以60%-90%乙醇-水系统进行梯度洗脱,共收集到三个组分c
1-1
、c
1-2
、c
1-3
。所得组分c
1-1
经凝胶柱色谱以60%甲醇-水进行等度洗脱,得到三个组c
1-1-1
、c
1-1-2
、c
1-1-3
。组分c
1-1-2
经硅胶柱
色谱以二氯甲烷-甲醇系统(50:1-1:1)进行梯度洗脱,经薄层检识和hplc分析进行合并得到五个组分c
1-1-2-1-c
1-1-2-5
;所得组分c
1-1-2-2
经ods柱色谱以20%-90%甲醇-水进行梯度洗脱,得到六个组分c
1-1-2-2-1-c
1-1-2-2-6
。利用半制备反相hplc对组分c
1-1-2-2-5
以乙腈-水系统(25:75)进行洗脱,得到了一对差向异构体1(12.3g)和2(10.2g)。利用daicel chiralpakad-h和daicel chiralpak ic手性色谱柱,流动相为正己烷-异丙醇混合溶剂,比例分别为8:1和5:1(v:v),流速分别为2.00和0.45ml/min,紫外检测器检测波长为210nm。对该对差向异构体1和2进行手性拆分得到化合物1a-1d和2a-2d。
[0048]
实施例2:经化合物1a-1d和2a-2d对体外adp诱导的血小板聚集抑制作用的考察。
[0049]
(1)血小板聚集实验步骤:
[0050]
1、3.8%柠檬酸钠的配制(现用现配)
[0051]
取柠檬酸钠0.19g,加蒸馏水定容至5ml,溶解后过滤,装瓶,121℃高压灭15min。
[0052]
2、药物的配制:
[0053]
(1)药物配制:称取适量样品,用生理盐水配置浓度在0.25mg/ml,dmso含量5%。
[0054]
(2)阴性对照:量取dmso 20μl,用生理盐水配置成5%浓度的dmso溶液。
[0055]
(3)阳性对照:称取阿司匹林1.0mg,生理盐水配置成0.25mg/ml的溶液,dmso含量5%。
[0056]
3、adp的配制
[0057]
称adp 5.551mg用生理盐水定容至5ml,混匀后分装,在5个1.5ml的ep每1ml,-20℃保存,现用现融,用的时候将其稀释10倍,用完再放到-20℃保存,溶解时不停颠倒混匀即可。将血液注入可离心的塑料管中,再加入抗凝剂,使血液:抗凝剂=9:1,轻轻的摇匀,忌用力震荡。
[0058]
4、富血小板血浆(prp)制备:抗凝血标本在200
×
g(1700转/分),离心8分钟,待离心机自然停止后取出标本,小心吸取上层富含血小板血浆。(离心后的prp中不应含有红细胞,否则应再离心,应根据所用离心机调整合适的转速及离心时间(以prp得量占血样1/3为佳)。prp应无溶血。
[0059]
贫血小板血浆(ppp)制备:将上述已吸取prp的血再次离心。1500
×
g(3900转/分),离心10分钟,待离心机自然停止后取出标本,小心吸取上层贫血小板的血浆备用,血浆应清亮透明。
[0060]
富血小板血浆中血小板数量调整,使血小板计数为4
×
105/mm3左右。
[0061]
5、精密量取ppp 400μl加入测试杯,进行仪器调零;另取一个测试杯,精密加入prp 125μl,然后再精密加入供试样品溶液100μl,完成后,将测试杯置于测试孔中,37℃温育1min,加入磁珠搅拌均匀,再加入25μl adp,迅速按下开始按钮,记录血小板聚集曲线,考察药物抑制血小板聚集的生物活性。
[0062]
抑制率=(空白血浆的最大聚集-供试品的最大聚集率)/空白血浆的最大聚集率
[0063]
(2)血小板聚集实验结果:
[0064]
实验结果显示,化合物1a,1c,2b,2d在0.25mg/ml浓度下均表现出显著的抑制血小板聚集作用,相当于阳性药的83.67
±
3.25%,对映异构体间并且表现出一定的立体选择性的活性差异。
[0065]
表2血小板聚集的抑制率[(n=3)0.25mg/ml]
[0066]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1